This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
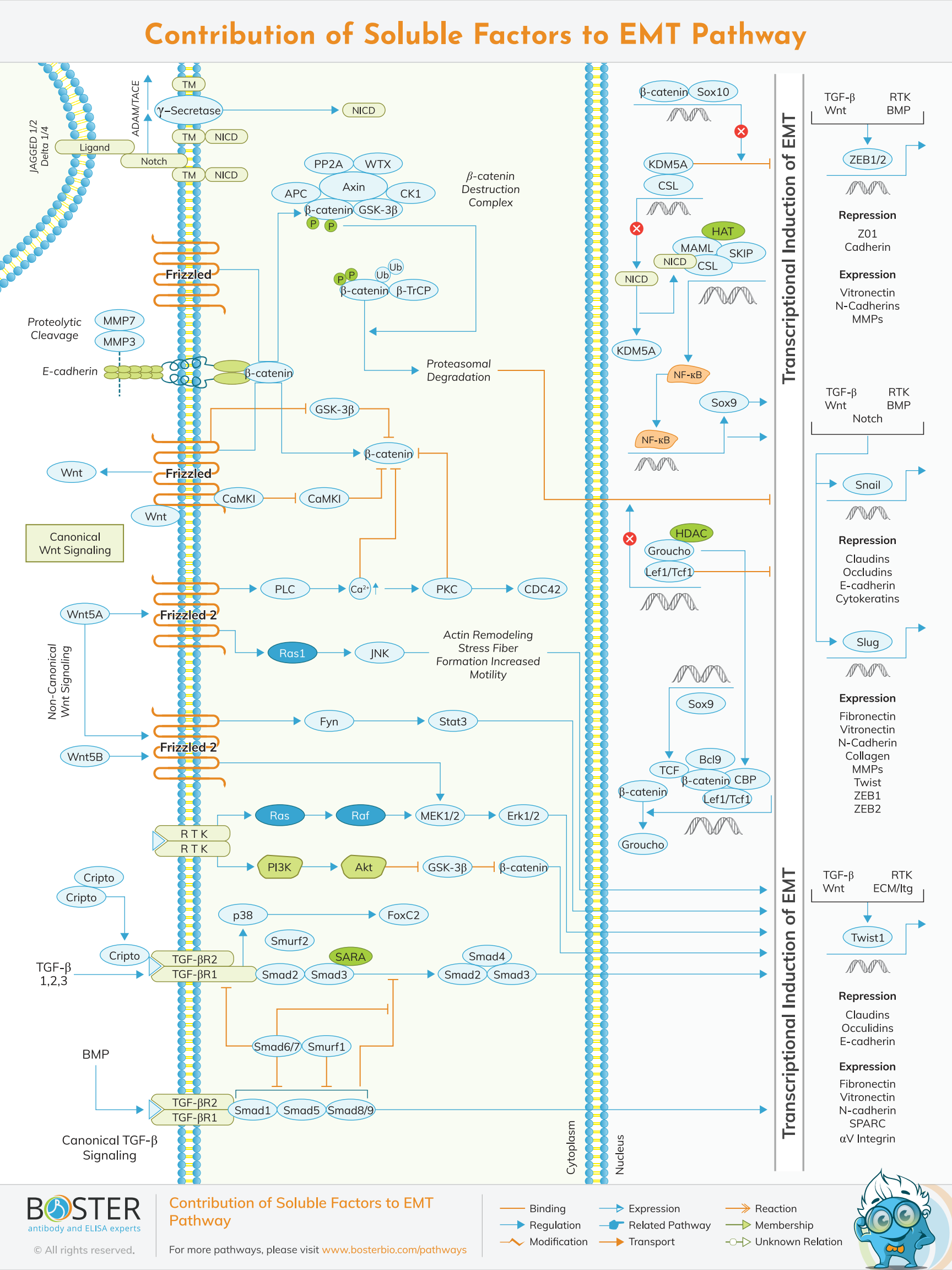
Numerous growth factors and cytokines can cause tumor cells to undergo an Epithelial-Mesenchymal Transition (EMT). These factors may be produced by tumor cells or stromal cells in the tumor microenvironment. These soluble ligands bind to their cognate receptors (e.g., tyrosine kinase receptors (RTKs), TGF- receptors), activating intracellular signaling pathways that promote EMT via upregulation of specific zinc finger (e.g., SNAI1, SLUG, ZEB1, ZEB2) or basic helix-loop-helix (bHLH, e.g., TWIST1) transcription factors.
It was first outlined as a feature of embryogenesis in 1980s by Betty Hay. Epithelial and mesenchymal cells differ phenotypically as well as function but they both share inherent plasticity. Epithelial are closely connected to each other by gap junctions, adherens and tight junctions, polarization of the action cytoskeleton have an apical-basal polarity and bound by a basal lamina. Mesenchymal cells in contrary lack this polarization. They have a spindle-shaped morphology and interact with each other through focal points only.
Despite paradoxically acting as a tumor suppressor in some contexts, the TGFβ signaling pathway is strongly implicated in the process of EMT induction. The canonical TGF-β/BMP signaling pathway is initiated when ligands (e.g., TGFβ1, TGF—β2, and BMPs) bind to their cognate Type I and Type II kinase receptors, forming a heterotetrameric ligand-receptor complex that recruits and phosphorylates receptor-regulated SMADs (R-SMADs). Phosphorylated R-SMADs (R-SMAD1/5/8 for BMP signaling; R-SMAD2/3 for TGF-β signaling) interact with a common mediator co-SMAD (SMAD4), causing the SMAD complex to translocate to the nucleus, where it regulates the expression of target genes involved in EMT (e.g., SNAIL, SLUG, etc.).
Additionally, TGF-beta-induced non-Smad signaling and cross-talk with other signaling pathways play critical roles in the induction of EMT. Ras signaling, in particular, interacts with TGF-beta-Smad signaling and plays a critical role in the induction of EMT.
TGF-beta inhibitors inhibit advanced cancer invasion and metastasis via a variety of mechanisms, including inhibition of EMT. The identification of molecules that inhibit TGF-beta-induced EMT but not growth arrest may be an ideal strategy for cancer invasion and metastasis treatment.
EMT can also be induced when non-canonical TGF-B bsignaling pathways are activated. For example, activation of the PI3K and Ras/Raf signaling pathways by oncogenic RTKs in conjunction with TGF- signaling has been considered a central feature of cancer EMT. The Ras/Raf pathway promotes transcriptional activation of key EMT promoter genes, whereas the PI3K pathway inhibits GSK3-mediated phosphorylation of -catenin, thereby allowing EMT target genes to be transcriptionally activated. Additionally, the TGFR activates p38 MAPK, which results in the activation of the EMT transcription factor FOXC2.
E-cadherin is a critical component of adherens junctions, which are responsible for epithelial integrity. EMT-associated transcription factors (e.g. SNAIL) suppress E-Cadherin expression while simultaneously inducing the expression of matrix metalloproteinases (MMPs) that degrade epithelial
E-Cadherin, resulting in the characteristic loss of epithelial integrity associated with EMT. E-cadherin degradation also results in the release of -catenin from adherens junctions, which can then translocate to the nucleus and activate oncogenic Wnt target genes.
Aberrant Wnt signaling is a hallmark of several cancers, most notably colorectal cancers, which overexpress -catenin in 90% of cases. Without Wnt signaling, free -catenin in the cytoplasm is rapidly phosphorylated and ubiquitinated by the -catenin destruction complex, preparing it for proteasomal degradation. When Wnt binds to the Frizzled receptor, GSK3 inhibits the -catenin destruction complex, and -catenin translocates to the nucleus, dislodging the Groucho/HDAC inhibition complex and activating TCF/LEF-mediated transcription of EMT effectors (e.g., SNAIL, N-Cadherin). EMT mediated by Wnt/-catenin has been demonstrated in a variety of cancers (osteosarcoma, gastric, prostate). Along with activating the expression of EMT effectors, LEF1 regulates the expression of microRNAs (miRNAs) involved in the promotion of EMT. Additional factors, such as SRY-Box 10 (SOX 10), regulate WNT/-catenin/LEF activity by competing with -catenin for binding to TCF/LEF.
Wnt5A and Wnt5B bind to the Frizzled2 (Fzd2) receptor and induce EMT by activating the non-canonical STAT and MEK/ERK signaling pathways, which have been shown to be upregulated in metastatic liver, lung, colon, and breast cancer cell lines and tumors. Additionally, Wnt5A/Frizzled signaling has been shown to induce EMT via non-canonical Wnt signaling pathways, including JNK signaling in pancreatic carcinoma and PKC signaling in melanoma, with Wnt5a expression increasing cellular motility in both pathways. Consistent with the increased motility, it has been demonstrated that WNT5a-mediated PKC signaling promotes filamin A expression and processing, which results in actin remodeling and stress fiber formation, both of which are required for motility in melanoma cell lines. I
In contrast to canonical Wnt signaling, Wnt5a expression is decreased in the majority of colon carcinoma tumors and cell lines and correlates negatively with EMT markers, whereas tumors with increased Wnt5A expression exhibit increased intracellular calcium and non-canonical Wnt signaling, decreased EMT, cellular motility, and invasiveness. It has been demonstrated that overexpression of Wnt5a in colon carcinoma cell lines with low basal levels of Wnt5A increases calcium ion levels and inhibits EMT marker expression, cellular motility, and proliferation while activating PKC and CaMKII activities, preventing -catenin nuclear localization, and abrogating EMT effector expression (e.g., TWIST, ZEB1).
NOTCH signaling is a juxtracine signaling pathway that is activated when ligands (Jagged 1/2, DLL1/3/4) expressed on the surface of adjacent cells bind to NOTCH receptors (NOTCH1-4). Ligand binding results in the ADAM/TACE protease cleavage of Notch, followed by -secretase cleavage and the release of the NOTCH intracellular domain (NICD), which translocates to the nucleus and regulates the transcription of downstream target genes. NICD displaces the transcriptional repressor KDM5A from a transcription factor complex in the nucleus, thereby promoting the expression of target genes involved in EMT. While Notch signaling is most frequently associated with T cell cancers (e.g. T-ALL), a Notch-driven EMT-like process has been described in a subset of epithelial cancers, including prostate and pancreatic carcinomas.
While Notch signaling is most frequently associated with T cell cancers (e.g. T-ALL), a Notch-driven EMT-like process has been described in a subset of epithelial cancers, including prostate and pancreatic carcinomas.