This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Pro tips on resolving common Western Blot issues such as weak signal, wrong band size, smiley gel, and high background.
Western blotting is a powerful technique for protein detection, but it can present several technical challenges. Unexpected results such as weak signals, high background, or non-specific protein bands appearing at unusual molecular weights, sometimes caused by protein degradation products, can arise from issues at any stage of the workflow, from sample preparation to primary or secondary antibody incubation or detection reagents handling.
The following Western blot troubleshooting guide serves as a checklist for the possible causes and solutions to some of the most commonly encountered problems with Western blot assays, complementing the standard SDS-PAGE gel and various protein blotting methods used in Western blotting protocol. Whether you're dealing with inconsistent results or complete signal loss, use this checklist to systematically pinpoint the source of the issue and implement targeted solutions.
If you do not see the issues you are having featured in this page, please contact us at support@bosterbio.com and we will help you resolve your specific trouble.

Download troubleshooting handbooks for IHC, Western blot, and ELISA for FREE.
Troubleshooting GuidesLet Bosterbio handle your Western blot CRO services and be your long term partner in broadening the bandwidth of your lab/team in a flexible manner.
High background on a western blot occurs when the membrane background signal of the membrane reduces the signal-to-noise ratio to unreadable levels. This can result from protein aggregation, improper blocking, or cross-reactivity of antibody reagents.
Practical solutions include optimizing primary antibody and secondary concentrations, carefully adjusting secondary antibody incubations, testing different blocking agents, and ensuring that nitrocellulose membranes or PVDF membranes are selected according to experimental needs, since the extraction of membrane proteins can influence background levels depending on the method used. For chemiluminescence-based assays, using high-quality ECL reagents and a fresh chemiluminescent substrate helps minimize membrane background and improve detection of your protein target.
Use these tips to identify and resolve the source of your unexpected band sizes.

| S.No. | Possible Cause | Solution |
|---|---|---|
| 1 | Antibody concentration is too high |
|
| 2 | Aggregate secondary antibody formation |
|
| 3 | Too high antibody incubation temperature |
|
| 4 | Non-specific secondary antibody binding or cross-reactivity with blocking agent |
|
| 5 | Cross-reactivity of primary or secondary antibody with blocking agent |
|
| 6 | Incompatible blocking agent |
|
| 7 | Incomplete blocking |
|
| 8 | Insufficient blocking |
|
| 9 | Cross-reactivity of antibody with other proteins |
|
| 10 | Insufficient washing |
|
| 11 | Exposure time is too long |
|
| 12 | Membrane problem |
|
| 13 | Insufficient membrane wash |
|
| 14 | Incompatible membrane |
|
| 15 | Dry membrane |
|
| 16 | Contaminated buffer |
|
| 17 | Contaminated equipment |
|
| 18 | Insufficient antibody binding activity |
|
| 19 | Excessive substrate incubation |
|
| 20 | Blocking proteins reacting with detection system |
|
A weak signal is characterized by faint or indistinct protein bands. While the bands may be barely visible at their predicted sizes, weak signals can often require repeating the experiment. Common causes include incomplete protein transfer, insufficient loading buffer, or degraded samples where protein degradation products accumulate due to insufficient protease inhibitors or phosphatase inhibitors.
Use the tips below to identify the source of the error and get better results.
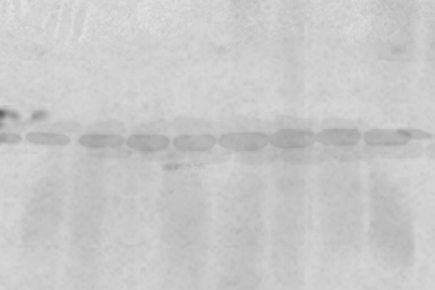
| S.No. | Possible Cause | Solution |
|---|---|---|
| 1 | Improper protein transfer to membrane |
|
| 2 | Insufficient protein and membrane binding |
|
| 3 | Insufficient antibody |
|
| 4 | Insufficient antigen |
|
| 5 | Antigen masking by blocking buffer |
|
| 6 | Presence of sodium azide in buffers |
|
| 7 | Too short exposure time |
|
| 8 | Too short substrate incubation time |
|
| 9 | Digestion of protein on membrane |
|
| 10 | Degradation of protein during storage |
|
| 11 | Incompatible primary and secondary antibodies |
|
| 12 | Low concentration of primary antibody and/or secondary antibody |
|
| 13 | Cross-reactivity between blocking agent and antibodies (primary or secondary) |
|
| 14 | Inability of primary antibody to recognize the protein in tested sample |
|
| 15 | Low or none content of target protein (ineffective antigen) |
|
| 16 | Insufficient transfer and excessive wash |
|
| 17 | Over-blocking |
|
| 18 | Loss of primary antibody effectiveness |
|
| 19 | Inhibition of secondary antibody by sodium azide |
|
| 20 | Loss of effectiveness in enzyme conjugate and substrate |
|
| 21 | Improper wet transfer for membrane |
|
| 22 | Insufficient molecular weight of target protein (< 10 kDa) |
|
| 23 | Equality or nearness in values between target protein’s isoelectric point and transfer buffer’s pH value |
|
| 24 | Too high methanol concentration |
|
| 25 | Insufficient sample concentration |
|
| 26 | Transfer too vigorous |
|
| 26 | Inadequate transfer |
|
| 27 | Sandwich assembly oriented incorrectly |
|
| 28 | Incorrect transfer buffer pH |
|
| 29 | Insufficient antibody binding affinity |
|
| 30 | Insufficient sample loading |
|
The non-specific background of a western blot does not always appear clean and flawless. Blotches, streaks, and spots are all common artifacts that can make it hard to interpret and publish your results. These artifacts are most commonly the result of uneven coating of buffer or antibody, the membrane drying out, or aggregates forming in the antibody or blocking buffer.
Follow the tips below to identify and solve the cause of your imperfect western blot background.
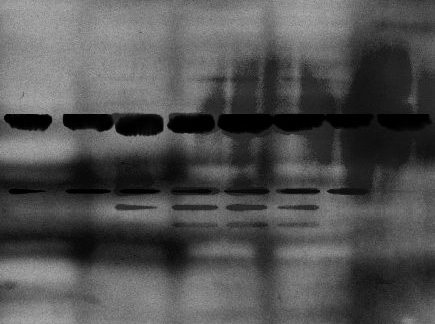
| S.No. | Possible Cause | Solution |
|---|---|---|
| 1 | Blotched background: Uneven antibody distribution |
|
| 2 | Blotched background: Membrane dried out unevenly |
|
| 3 | Blotched background: Uneven wash/incubation buffer coverage |
|
| 4 | Flecked background: Secondary antibody aggregation |
|
| 5 | Flecked background: Clumps of blocking buffer binding secondary antibody |
|
| 6 | Flecked background: Buffer contamination |
|
| 7 | White spots with no protein transfer: Air bubbles trapped between gel and membrane during transfer |
|
Misplaced or unexpected protein bands—appearing at incorrect molecular weights— may result from sample degradation, post-translational modifications, the presence of a splice variant or cross-reactivity of the primary antibody. Using stronger reducing agents, preparing fresh loading buffer, and validating results with different detection reagents or a new primary antibody can help confirm the source of error.
Use these tips to identify and resolve the source of your unexpected band sizes.
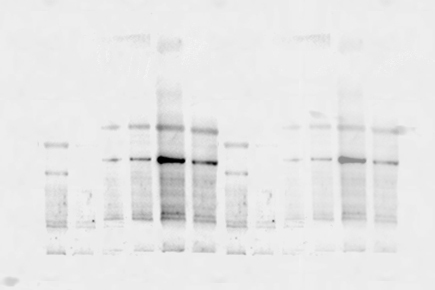
| S.No. | Possible Cause | Solution |
|---|---|---|
| 1 | Bands have higher MW than expected: Proteins are glycosylated or bear other post-translational modifications |
|
| 2 | Bands have much higher MW than expected: Protein aggregation |
|
| 3 | Bands have much higher MW than expected: Incomplete denaturation or residual disulfide bonding |
|
| 4 | Bands have lower MW than expected: Protein sample has been digested or degraded |
|
| 5 | Bands have lower MW than expected: Primary antibody is detecting splice variants |
|
| 6 | Bands have lower MW than expected: Primary antibody binding a similar epitope on a different protein |
|
| 7 | Multiple bands: Primary or secondary antibody contaminated with nonspecific IgG |
|
| 8 | Multiple bands: Nonspecific binding of primary antibody |
|
| 9 | Multiple bands: Nonspecific binding of secondary antibody |
|
| 10 | Multiple bands: Insufficient blocking |
|
| 11 | Multiple bands: Ionic interactions |
|
Distortions such as smile-shaped or streaked bands usually occur due to uneven running conditions in the SDS-PAGE gel or improper protein transfer.
Lowering the voltage, running gels at cooler temperatures, and ensuring no air bubbles are trapped between the gel and the membrane improves consistency. Careful alignment of nitrocellulose membranes or PVDF membranes is also critical for accurate protein detection.
Make sure your next blot has even, crisp bands by following the tips below.
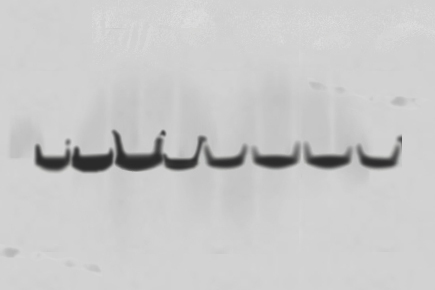
| S.No. | Possible Cause | Solution |
|---|---|---|
| 1 | Curved, "smiling" bands: Electrophoresis voltage too high |
|
| 2 | Curved, "smiling" bands: Overheated gel |
|
| 3 | Streaked or diffuse bands: Incomplete contact between gel and membrane during transfer |
|
| 4 | Streaked or diffuse bands: Slippage of membrane during transfer |
|
| 5 | Blurry bands: Electrophoresis voltage too high |
|
| 6 | Blurry bands: Improper loading buffer composition |
|
| 7 | Blurry bands: Air bubbles trapped between gel and membrane during transfer |
|
| 8 | Ghost bands: Overexposure during visualization |
|
| 9 | Ghost bands: Loading sample too concentrated |
|
| 10 | Ghost bands: Antibody concentration too high |
|
| 11 | Ghost bands: Blot was moved during transfer |
|
| 12 | Uneven, crooked bands: Poor gel polymerization |
|
| 13 | Uneven, crooked bands: Varying salt concentration between wells |
|
Maximize the value of your research and obtain critical insights from your samples through our specialized Western blotting service. Contact us now to discuss your project requirements and see how our expert Western blotting service can support your research.

Learn the concept behind Western blotting. It is a technique that is used to detect specific proteins in the given sample. It usually involves two major processes, namely, SDS-polyacrylamide gel electrophoresis and protein blotting and testing.
Learn about Western Blot Principle
Check out this Western Blot sample preparation guides to learn how to optimize every step, from improving protein yield to buffer preparation, to get the best results from your sample type. Learn more about sample preparation in this guide.
Learn Western Blot Sample Preparation
Learn a stepwise Western blotting protocol from reagent preparation to detection with application of BosterBio reagents. Check out our ELISA protocols to learn how to get the best results.
Check our Western Blot Protocol
Get to learn the concept behind our best practises on Western Blot optimization. Learn how to optimize every aspect of your experiment to yield the best results.
Browse Western Blot Optimization Tips