This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Explore the fundamentals and workflow of Western blotting—from core principles to detailed protocols—all supported by Boster Bio’s trusted reagents and CRO expertise.
Check out what Western Blot reagents we offerWestern blotting, also known as protein immunoblotting, is a widely used method for detecting specific proteins in complex biological samples following extraction with a lysis buffer supplemented with protease inhibitors to prevent protein degradation. It combines gel electrophoresis with antibody-based detection to identify target proteins with high specificity and sensitivity.
The process begins with SDS-polyacrylamide gel electrophoresis (SDS-PAGE), which separates proteins based on size. These proteins are then transferred to a membrane—typically nitrocellulose membranes or PVDF membranes, where they are probed using antibodies specific to the protein of interest. The resulting signal reveals both the presence and relative abundance of the target protein. This approach allows researchers to confirm protein extraction , examine post-translational modifications, and validate data from complementary assays such as ELISA or PCR.
Western blotting is a foundational technique in molecular biology, biochemistry, immunogenetics, and clinical diagnostics. It is capable of detecting proteins at concentrations as low as 1 ng, making it ideal for sensitive protein-level analysis in cells, tissues, or biofluids.
Boster Bio supports your Western blotting workflow with validated antibodies, high-performance reagents, and expert CRO services. Whether you're optimizing a protocol or scaling up your studies, we provide the tools and guidance to help you generate consistent, reproducible results.
You can Save up to 90% on WB reagents if you buy them from Boster Bio
Western blotting principle usually involves two major processes, namely, SDS-polyacrylamide gel electrophoresis and protein blotting and testing.
The electrostatic repulsion that is created by binding of SDS causes proteins to unfold into a rod-like shape thereby eliminating differences in shape as a factor for separation in the gel. Minor axis of all rods, the SDS-protein subunit compound are nearly the same, about 1.8nm. And the length of major axis is in proportion to molecular weight of the protein subunit. Thus electrophoretic mobility of the SDS-protein subunit compound is based on molecular weight, eliminating the influence imposed by size and charge.
The sample to be analyzed is mixed with SDS. And the mixed samples are subsequently treated by related solution. Heating the samples to at least 60°C further promotes protein denaturation and depolymerization, helping SDS to bind and enabling the rod-shape formation and negative charge adherence. A bromophenol blue dye may be added to the protein solution to allow the experimenter to track the progress of the protein solution through the gel during the electrophoretic run. An appropriate amount of glycerol is added to increase density and accelerate the migration of sample solution.
Selecting the appropriate gel percentage is essential for accurate protein separation in SDS-PAGE. The gel’s acrylamide concentration determines its pore size, directly influencing the resolution of proteins based on their molecular weight. This section explains how to choose the right gel percentage for your target protein, includes a calculator for convenience, and outlines the relationship between gel composition and pore size.
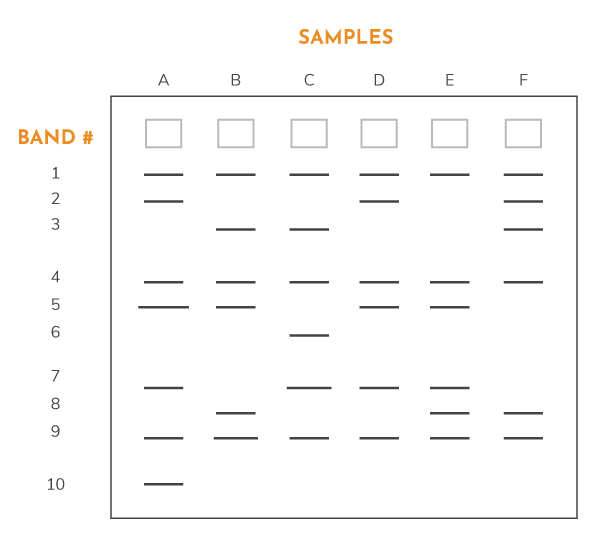
Polyacrylamide gel electrophoresis (PAGE) is used for separating proteins ranging in size from 5 to 2,000 kDa due to the uniform pore size provided by the polyacrylamide gel. Pore size is controlled by controlling the concentrations of acrylamide and bis-acrylamide powder used in creating a gel. Typically resolving gels are made in 5%, 8%, 10%, 12% or 15%. Stacking gel (5%) is poured on top of the resolving gel and a gel comb (which forms the wells and defines the lanes where proteins, sample buffer and ladders will be placed) is inserted. The percentage chosen depends on the size of the protein that one wishes to identify or probe in the sample. The smaller the known weight, the higher the percentage that should be used. Changes on the buffer system of the gel can help to further resolve proteins of very small sizes
| Range of molecular weight (KD) | Concentration of gel (%) |
|---|---|
| <10 | 15 |
| 10 - 30 | 12 |
| 30 - 100 | 10 |
| 100 - 500 | 8 |
| > 500 | 5 |
Five steps are involved in western blotting procedure and detection assay, namely, protein transfer, blocking, primary antibody incubation, secondary antibody incubation and protein detection, and western blotting analysis—each contributing to the reliability of your protein analysis.
Proteins are moved from within the gel onto a nitrocellulose membranes (NC) or polyvinylidene difluoride (PVDF) using a technique known as electrophoretic transfer. Without pre-activation, proteins combine with nitrocellulose membrane based on hydrophobic interaction, thereby having slight effect on protein activities. Besides, nitrocellulose membrane produces little non-specific staining. It is cheap and ease to use. However, it is easy to erase small molecular proteins while washing. It is fragile and has poor toughness. With high affinity, the PVDF membrane needs to be sunk in methanol before use to activate positive charge groups on the membrane, promoting combination with negative charged proteins. Specific NC membrane with different pores should be applied according to the molecular weight of transferred proteins due to the smaller the pore of membrane the tighter the combination between membrane and small molecular weight proteins. NC membranes of 0.45 µm and of 0.2 µm are used most. The size of 0.45 µm should be applied for proteins with molecular weight over 20KD while the size of 0.2 µm will be chosen for those below 20KD. PVDF membrane is best for the detection of small molecular weight proteins due to its higher sensitivity, resolution as well as affinity than normal membrane.
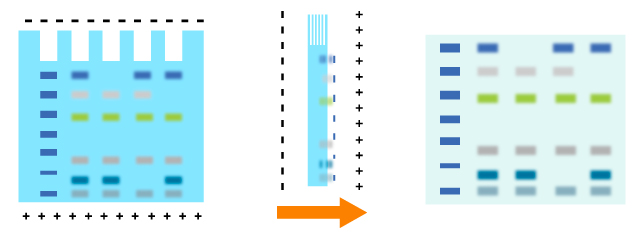
Transfer methods that are used most for proteins are semi-dry transfer and wet transfer which rely on a transfer buffer to facilitate protein transfer. Semi-dry transfer describes the method that Gel-Membrane-Filter sandwich is placed between filters loaded with transfer buffer. The transfer process is based on current conduction produced by the transfer buffer. Semi-dry transfer takes little time with high efficiency as electric current works directly on membrane and gel. While applying wet transfer, the Gel-Membrane-Filter sandwich is placed in the transfer tank, suspending in transfer buffer vertically. Proteins transfer from the gel to the membrane under the control of high intensity electric field produced by electrode plate paralleled to the sandwich. While prolonging time to an appropriate extend, proteins could be transferred more effectively. Proteins within several gels could be transferred.
In a western blot, it is important to block the unreacted sites on the membrane using blocking solutions to reduce the amount of nonspecific binding of proteins during subsequent steps in the assay using inert protein or nonionic detergent. Blocking buffers should block all unreacted sites. And Blocking buffers should not replace target protein on the membrane, not bind epitope on the target protein and not cross react with antibody or detection reagents. The most typical blocking agents are BSA, nonfat dry milk, casein, gelatin and Tween-20. TBS and/or PBS are the most commonly used buffers.

Inertia protein BSA, nonfat dry milk, casein, gelatin or nonionic detergent Tween-20 reduce nonspecific binding by blocking unreacted sites. Retaining protein structure, Tween-20 can reduce breakup to original interaction among proteins while is used for protein emulsification.
After blocking, primary antibody specific to target protein is incubated with the membrane. And the primary antibody binds to target protein on the membrane.
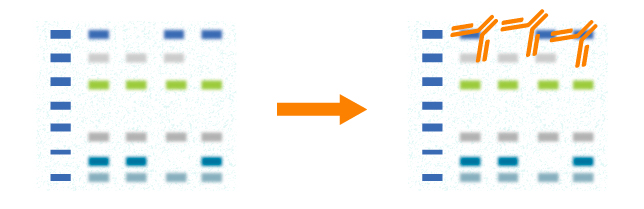
In western blot, primary antibody should be validated before use. The choice of a primary antibody depends on the antigen to be detected. Both polyclonal and monoclonal antibodies work well for western blot. Monoclonal antibodies recognize single specific antigenic epitope. Thus, they have higher specificity resulting in lower background. Blot results will be influenced if the target epitope is destroyed. Polyclonal antibodies recognize more epitopes and they often have higher affinity. Blot results will be stable even though a few epitopes are destroyed.
After rinsing the membrane to remove unbound primary antibody, the membrane is exposed to a specific enzyme-conjugated secondary antibody. And the secondary antibody binds to the primary antibody which has reacted with the target protein.

The most popular secondary antibodies are anti-mouse and anti-rabbit immune globulin since the host species for primary antibodies are mainly mouse and rabbit. Goat is used widely to raise anti-mouse and anti-rabbit polyclonal antibodies. Thus, goat anti-mouse and goat anti-rabbit immune globulin are the most commonly used secondary antibodies. The choice of secondary antibody depends upon the species of animal in which the primary antibody was raised. For example, if the primary antibody is a mouse monoclonal antibody, the secondary antibody must be an anti-mouse antibody. If the primary antibody is a rabbit polyclonal antibody, the secondary antibody must be an anti-rabbit antibody.
Protein detection (color development) and analysis of Protein detection (color development)A substrate reacts with the enzyme that is bound to the secondary antibody to generate colored substance, namely, visible protein bands. The target protein levels in cells or tissues are evaluated through densitometry and the location of the visible protein bands.
Alkaline phosphatase (AP) and horseradish peroxidase (HRP) are two enzymes commonly employed in Western blot detection systems. Functioned by Alkaline phosphatase (AP) catalyzation, a colorless substrate BCIP will be converted to a blue product. In the presence of H2O2, 3-amino-9- ethyl carbazole and 4-chlorine naphthol will be oxidized into brown substance and blue products respectively under the catalyzation of HRP. Enhanced chemiluminescence is another method that employs HPR detection. Using HRP as the enzyme label, luminescent substance luminol will be oxidized by H2O2 and will luminesce. Moreover, enhancers in this substrate will enable a 1000-fold increase in light intensity. HRP will be detected when the blot is sensitized on photographic film.
After color development, the pattern of the separated proteins is imprinted onto the film or captured by Western Blot gel imager. By comparing the band position to the protein ladder, one can estimate the size of the protein. Some innovative WB imagers can be found at Azure Biosystems.
The reagents you will need for each step are listed below.

Proper control design is essential to ensure the accuracy, specificity, and reproducibility of Western blot results. Controls help validate antibody performance, confirm protein expression, and identify sources of background noise or technical error. Without controls, experimental findings lack scientific reliability.
Proper control design is essential to western blot. It will guarantee accurate and specific test result by identifying various problems quickly and precisely. There are 5 common types of controls seen in Western blot experiment design.
Loading controls are required to check that the lanes in your gel have been evenly loaded with sample, especially when a comparison must be made between the expression levels of a protein in different samples. They are also useful to check for even transfer from the gel to the membrane across the whole gel. Where even loading or transfer have not occurred, the loading control bands can be used to quantify the protein amounts in each lane. For publication-quality work, use of a loading control is absolutely essential
| Loading Control | Molecular Weight (KD) | Sample Type |
|---|---|---|
| Beta-actin | 43KD | Whole Cytoplasmic |
| GAPDH | 30 - 40KD | Whole Cytoplasmic |
| Tubulin | 55KD | Whole Cytoplasmic |
| VCDA1/Porin | 43KD | Mitochondrial |
| COXIV | 16KD | Mitochondrial |
| Lamin B1 | 16KD | Nuclear (Not suitable for samples where the nuclear envelope is removed.) |
| TBP | 38KD | Nuclear (Not suitable for samples where the nuclear envelope is removed.) |
Sample preparation, protocol, troubleshooting and more.
The choice of protein extraction method is crucial. It ultimately makes the difference between a blank blot and a beautiful one.
WB sample preparation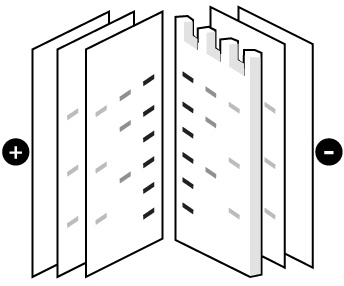
A step-wise guide of optimized WB protocol. Best choice for setting up a new SOP for your lab or for educating new lab members.
WB protocol
Something wrong with your blot? No worries, check out the comprehensive troubleshooting guide to see if your issue is already covered.
WB troubleshooting