This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
We have a convenient Plasmid DNA Extraction and Purification MiniPrep Kit for your molecular biological experiments.
Check it outOn this page, we will cover the protocols for:

Download this comprehensive manual which discusses the basic principles and techniques of PCR and molecular biology research, including protocols and troubleshooting solutions.
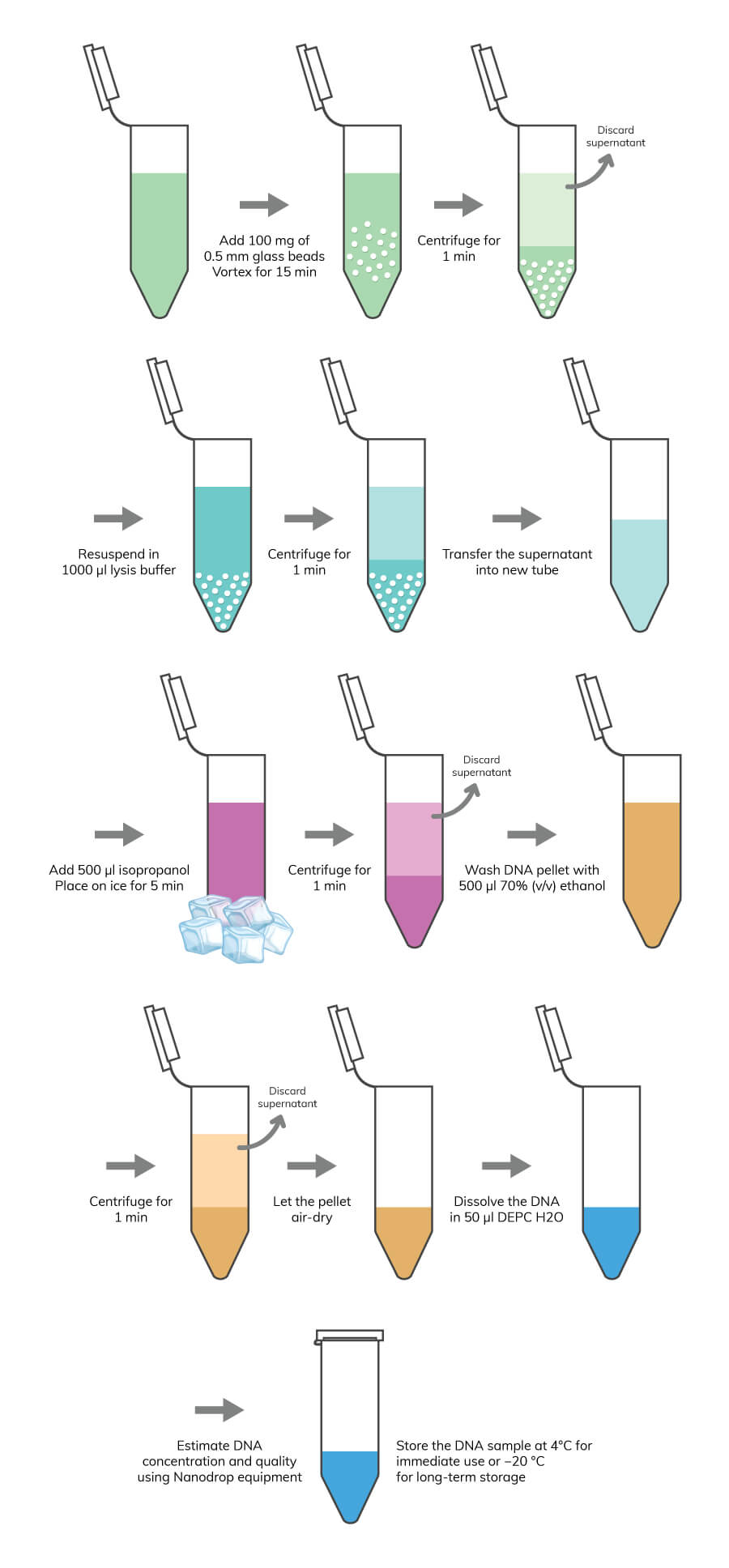
Download PCR eBookThe genomic DNA (gDNA) and total RNA are frequently the genetic elements targeted for molecular biology experiments since these are the main sources of genetic information in an organism.
Genomic DNA and total RNA extraction have a straightforward methodology, requiring only cell lysis to accomplish the task. However, it is recommended to consider the type of organisms you are using in your lab research. Some organism’s cells have a cell wall structure, which confers extra protection to cells. This is the case for most yeast, plants, and bacteria species. For these organisms, it is recommended to include an extra step for cell lysis before disrupting the plasma membrane. This step is usually an enzymatic or mechanical process to disrupt the cell wall. The purification of DNA and RNA molecules is required to avoid contaminations with other intracellular components, such as proteins and metabolites. Some commonly used methods for DNA and RNA purification are precipitation with phenol-chloroform or isopropanol, or by spin columns with silica membrane.
If your research is focused on gene expression analysis, then you should target total RNA. The same procedure used to extract and purify gDNA can be used for total RNA purification, although some additional steps are required, as you will see in the following RNA extraction and purification protocol. Boster recommends the following protocols for sample preparation depending on your initial sample source.
When your target DNA is a plasmid, you should adapt your procedure to make sure the plasmid DNA (pDNA) is separated from gDNA. Alkaline lysis is the most current protocol used in the labs. The lysis buffer has in its composition sodium hydroxide and SDS, which will be responsible for denaturation of gDNA and pDNA. Subsequently, a neutralization buffer is added to renature the pDNA but not the gDNA. Because of the size of gDNA, it is much more difficult to renature again, unlike the pDNA. Then the same procedure of gDNA purification can be used for pDNA purification.
Protocol:
Note: If you are using cell suspensions, you can move directly to the lysis procedure. If you are using samples, like cell tissues that are difficult to homogenize, you can use liquid nitrogen to create a homogeneous cell suspension. At the time you have a cell suspension, you should be careful about the membrane structure of your sample. Usually, for cells with cell walls, a pre-lysis treatment is recommended to degrade this barrier. You can use an enzymatic method or a mechanical method.
Note: Alternatively, you can use 10U of lyticase (for fungi samples) or 10U of lysozyme (for gram-negative bacteria).
Note: Nucleic acids absorb UV light at 260 nm, whereas proteins and phenolic compounds absorb at 280 nm. Many organic compounds as well as phenol, TRIzol, and chaotropic salts have strong absorbances at around 230 nm. The A260/280 ratio is used to identify protein contamination. For RNA and DNA, A260/280 ratios should be around 2.1 and 1.8, respectively. The A260/230 ratio indicates the presence of organic contaminants, such as phenol, TRIzol, chaotropic salts and other aromatic compounds. Samples with 260/230 ratios below 1.8 have a significant amount of these contaminants.
Fast and simple plasmid miniprep method for routine molecular biology laboratory applications.
Protocol:
During this protocol, always use new gloves and RNase free material.
A simple spin column technique for preparation of high quality, high‐purity intact total RNA.
If the goal is to create a genomic DNA library, the first step is to extract genomic DNA (please see the protocol for DNA extraction). If the first step to generate a cDNA library relies on mRNA extraction, please see the protocol for RNA extraction. Afterwards, the mRNA is converted to cDNA through the catalytic activity of reverse transcriptase enzyme.
Once the cDNA is obtained, the use of restriction enzymes is required to create complementary ends in the vector and in the DNA fragments.
To functionally characterize and identify new genes, physiological tests can be carried out to detect phenotypic differences between the host organism and the host organism carrying DNA fragments of the DNA library. Usually growth tests are the most common approach to achieve this task, either by cultivating cells in different media formulations and/or different temperatures.
One real-life example is the use of renewable carbon sources aiming to produce biofuels as a recent initiative to find alternatives to fossil fuels. Most of the organisms used in this bioprocess, like E. coli, are not able to grow in these substrates, namely xylan. But the degradation product of xylan, which is xylose, can be assimilated and metabolized by industrial microbes (and used to produce biofuels). Thus, samples of microbes growing in substrates rich in xylan, were used to extract the RNA and prepare cDNA libraries as described previously, and to be transformed in E. coli competent cells. The transformed cells are subsequently plated in minimal media with xylan as sole carbon sources. This procedure will allow positive selection of transformants. Only transformants that carry genes in the vector that code for enzymes able to degrade xylan into xylose will grow in the plate. The vector of the transformants is then extracted and sequenced. This will allow us to identify which genes are responsible to allow growth in xylan media.
You can access more information about sample preparation, protocols, and troubleshooting in our PCR Technical Resource Center.