This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Boster Bio protocols for flow cytometry offer a step-by-step overview of the procedure. Use this guide as a primer or a quick reference guide, and see our product datasheets or sample preparation guides for more details.
Flow cytometry is a laser-based technique used to measure physical and chemical characteristics of cells or particles in suspension. It enables researchers to assess cell surface antigen expression, intracellular proteins, DNA content, and functional states through the use of fluorochrome-conjugated antibodies. A subtype of flow cytometry, fluorescence-activated cell sorting (FACS), allows for the physical separation of cell populations based on these markers.
Each human cell expresses hundreds of thousands of cell surface antigens that specify their cell type, biological function, development stage, and much more. Cells residing in different organs have characteristic cell surface antigens, and the determination of these cells using the specific fluorochrome-conjugated antibodies can be analyzed by flow cytometry. Staining with an unconjugated purified antibody needs an additional step of staining with a fluorescent conjugated secondary antibody (indirect immunostaining).
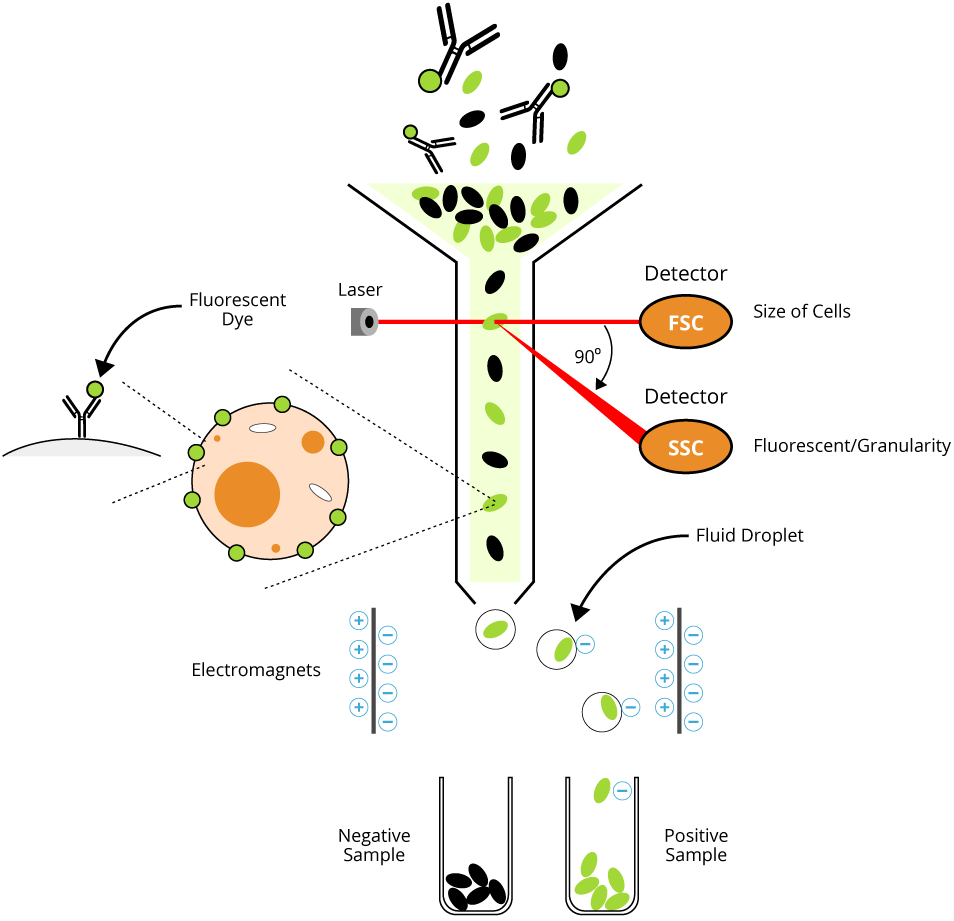
This information is to serve as a guide as individual investigators may need to optimize protocols for their particular cell type.
If the cells are stained in a 96 well U- or V-bottom plate, washing procedure should be set up first for maximum removal of unbound primary antibodies.
This guide will show you all the nuts and bolts for Flow Cytometry and FACS, including expert review of principle, optimized protocol that really works, and more.
These protocols cover staining of surface and intracellular antigens, including cytokines and phospho-proteins. Optimization is advised for each specific cell type and antibody panel.
Key Reagents – PBS, staining buffer, FACS buffer, PFA fixing buffer
Key Reagents – PBS, staining buffer, FACS buffer, PFAfixing buffer
Key Reagents – PBS, staining buffer, FACS buffer, 0.5-4% PFA in PBS [exact concentration of PFA has to be standardized for every antibody panel], 100% methanol
Key Reagents – PBS, staining buffer, FACS buffer, 0.5-4% PFA in PBS [exact concentration of PFA has to be standardized for every antibody panel], 0.1% saponin
Key Reagents – PBS, staining buffer, FACS buffer, 0.5-4% PFA in PBS [exact concentration of PFA has to be standardized for every antibody panel], 100% methanol
The intracellular staining procedure allows direct measurement of antigens (cytokines or transcription factors) present inside the cytoplasm or in the nucleus of a cell in addition to the surface antigen determination simultaneously. In this procedure, the fixation and permeabilization of cells are required after staining with fluorescently conjugated surface antigens. This modified staining procedure allows direct measurement of functional activity of any cell of interest present in the blood or other tissues without further separation. To achieve better results, additional in vitro cell stimulation with some common mitogen (e.g., PMA, Ca++ or peptide epitopes and protein transport inhibitor, Brefeldin A, etc.) may be required, which allows increased production of cytokines inside cells. Refer to the table below as a guideline for common cell stimulation procedures.
Note: If the cells are stained in a 96 well U- or V-bottom plate, washing procedure should be set up first for maximum removal of unbound primary or secondary antibodies.
| Target cytokine/phosphoprotein | Target cells | Stimulant | Duration | Surface marker |
|---|---|---|---|---|
| IL-2 | PBMCs | PMA (50ng/ml) | 4-6 hours | CD3 |
| IL-3 | T-cells | PMA(50ng/ml) + ionomycin (1µg(ml) | 4-6 hours | CD4 |
| IL-4 | PBMCs | PMA(50ng/ml) + ionomycin (1µg(ml) | 4-6 hours | CD4 |
| IL-6 | PBMCs | LPS (100ng/ml) | 4-6 hours | CD14 |
| IL-10 | PBMCs | LPS (100ng/ml) | 18-24 hours | CD14 |
| GM-CSF /IFNγ/TNFα/TNFβ | PBMCs | PMA(50ng/ml) + ionomycin (1µg(ml) | 4-6 hours | CD3 |
| pStat5 | PBMCs | GM-CSF (20ng/ml) + IL3 (20ng/ml) | 15 min. | CD123, CD116 |
| pStat3 | PBMCs | G-CSF (20ng/ml) + IL6 (20ng/ml) | 15 min. | CD126, CD114 |
| pERK | PBMCs | IL3 (20ng/ml) + IL6 (20ng/ml) + FLT3L (20ng/ml) | 15 min. | CD123, CD126, CD135 |
Dye exclusion staining is performed to separate live and dead cells, as well as to isolate the rare stem cell ‘side populations’. If viability staining is included in your regular immunostaining, it should be performed before any other staining.
Key reagents – PBS, staining buffer, PI solution (10µg/ml in PBS)
Key reagents – PBS, staining buffer, 7-AAD solution (100µg/ml in PBS)
Key reagents – PBS, 5% FBS in PBS, staining buffer, Hoechst 33342 solution (1mM in PBS) or Rho123 solution (10µg/ml in PBS)
Key reagents – PBS, staining buffer, PI solution (50µg/ml in PBS), RNAse A (10µg/ml), 70% ethanol
Learn the fundamental principle of flow cytometry and FACS, including how cells are counted, profiled, and sorted in a fluid mixture.
Identify common flow cytometry issues such as high background, weak signal, and poor population separation with practical troubleshooting tips.
Learn how to prepare cells and samples for flow cytometric analysis to support cleaner data and more reliable results.
Explore optimization tips for panel setup, staining conditions, and signal quality to improve overall flow cytometry performance.
Improve sample quality with practical guidance on cell handling, viability, and preparation methods before flow cytometry analysis.
Optimize fixation and permeabilization methods for intracellular staining to improve consistency and preserve target detection.
Learn how to optimize fluorescence staining, including antibody titration strategies that help balance signal strength and background control.
Review cell stimulation considerations for flow cytometry, including conditions relevant to cytokine detection and intracellular staining workflows.
Learn how cell-free antibody synthesis works and explore its potential advantages for rapid antibody generation and screening workflows.