This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Inhibitor of apoptosis (IAP) proteins are involved in cell death regulation in a variety of ways, from apoptosis and necrosis suppression to cell cycle and inflammatory regulation.
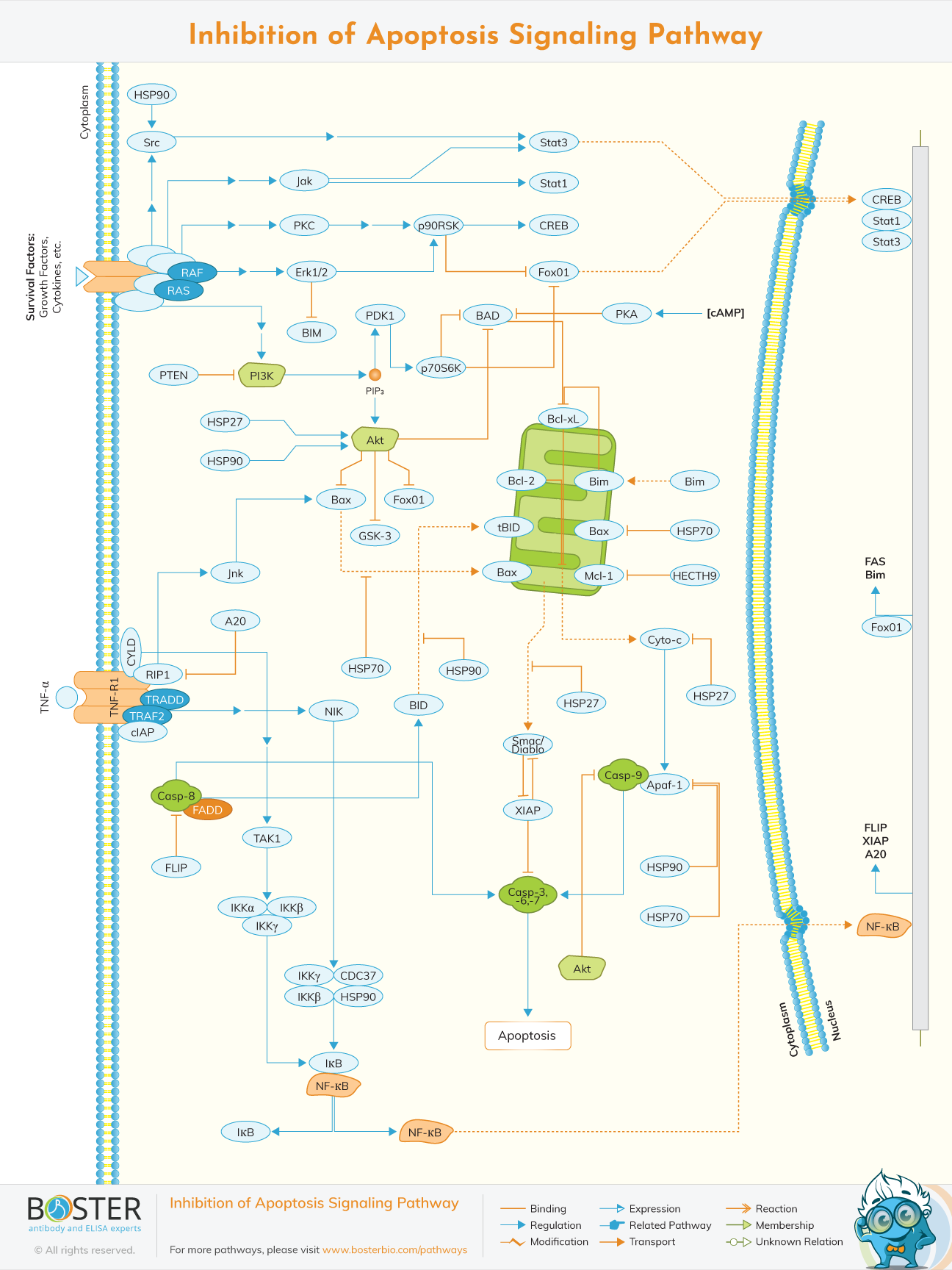
Cell apoptosis, sometimes called programmed cell death, is a cellular self-destruction method to remove old and damaged cells during development and aging to protect cells from external disturbances and maintain homeostasis. Apoptosis also occurs as a defense mechanism such as in immune reactions or when cells are damaged by disease or noxious agents.
Abnormalities in cell apoptosis can be a significant component of diseases such as cancer, autoimmune lymphoproliferative syndrome, AIDS, ischemia, and neurode-generative diseases. These diseases may benefit from artificially inhibiting or activating apoptosis. A short list of potential methods of anti-apoptotic therapy includes stimulation of the IAP (inhibitors of apoptosis proteins) family of proteins, caspase inhibition, PARP (poly [ADP-ribose] polymerase) inhibition, stimulation of the PKB/Akt (protein kinase B) pathway, and inhibition of Bcl-2 proteins
Ferroptosis and necroptosis are recently recognized forms of regulated cell death that differs considerably from apoptosis. Misregulated ferroptosis or necroptosis have also been implicated in multiple physiological and pathological processes, including cancer cell death, neurotoxicity, neurodegenerative diseases.
The intrinsic apoptosis pathway is initiated by, for example, chemotherapy and/or radiotherapy. It is activated by a range of exogenous and endogenous stimuli, such as DNA damage, ischemia, and oxidative stress. Moreover, it plays an important function in development and in the elimination of damaged cells.
In the intrinsic pathway, the functional consequence of pro-apoptotic signaling is mitochondrial membrane perturbation and release of cytochrome c in the cytoplasm, where it forms a complex or apoptosome with apoptotic protease activating factor 1 (APAF1) and the inactive form of caspase-9. This complex hydrolyzes adenosine triphosphate to cleave and activate caspase-9. The initiator caspase-9 then cleaves and activates the executioner caspases-3/6/7, resulting in cell apoptosis. It's totally different from the extracellular signals, which are usually generated by cytotoxic cells of the immune system and trigger apoptosis mainly through the extrinsic pathway.
The intrinsic apoptosis pathway induces apoptosis by directly activating caspase-3 or by cleaving bid (BH3 interacting domain death agonist), resulting in mitochondrial dysfunction and subsequent release of cytochrome c and activation of caspases-9 and caspases-3. Caspase-3 promotes the typical apoptosis features, including DNA fragmentation and cell death in several tissues.
The B-cell lymphoma 2 (Bcl-2) protein family tightly controls activation of the intrinsic pathway. It is found in follicular lymphoma and first identified as one of the genes involved in the cell death by either activating pro-apoptotic or inhibiting anti-apoptotic apoptosis. Proteins in one subgroup, including Bid, Bad, Bim, Bmf, Puma, and Noxa, contain a single Bcl-2 homology 3 domain (BH3-only proteins) and have pro-apoptotic activity. Two other protein subsets have multiple BH domains. The first subset, including Bcl-2 associated X protein (Bax), Bcl-2 homologous antagonist/killer (Bak) and Bcl-2 family apoptosis regulator (Bok), is pro-apoptotic; the other subset, including Bcl-2, Bcl-XL, and Mcl-1, is anti-apoptotic. The mitochondrial pathway is partly influenced by Bcl family members bound to the mitochondrial membrane, including both pro-apoptotic regulatory proteins Bax and anti-apoptotic regulatory proteins Bcl-2.
Apoptosis is known as a physiological process of cell deletion and is also a process of programmed cell death, resulting in morphological change and DNA fragmentation. It is stimulated by external or internal events of cells, one of which is the extrinsic pathway mediated by the death receptor. The death receptors include Fas receptors, tumor necrosis factor (TNF) receptors, and TNF-related apoptosis-inducing ligand (TRAIL) receptors. As a surface receptor, for example, TNF receptor-1 (TNF-R1), it will interact with TNF to induce the recruitment of adaptor proteins such as Fas-associated protein with death domain (FADD) and Tumor necrosis factor receptor type 1-associated DEATH domain protein (TRADD), which recruits a series of downstream factors, including Caspase-8, which is a critical mediator of the extrinsic pathway, resulting eventually in cell apoptosis.
The extrinsic pathway that initiates apoptosis is triggered by a death ligand binding to a death receptor, such as TNF-α to TNFR1. The TNFR family is a large family consisting of 29 transmembrane receptor proteins, organized in homotrimers and activated by binding of the respective ligand(s). They share similar cysteine-rich extracellular domains and have a cytoplasmic domain of about 80 amino acids called the "death domain" (DD). This death domain plays a critical role in transmitting the death signal from the cell surface to the intracellular signaling pathways. There are 19 members of the TNF ligand family and binding may result in a number of responses, including proliferation, inflammation, and apoptosis, depending on the adaptor proteins associated with the activated receptor.
TNFR may also stimulate pro-inflammatory pathways leading to activation of NFκB, via recruitment of RIP. The death domain kinase RIP is essential for TRAIL-induced IκB kinase (IKK) activation. It has been identified that the binding of TNF-α and TNFR1 activates NFkB pathway, which favored both cell survival and apoptosis, depending on the cell type and biological context.
Besides TNFR1, the Fas and DR4/DR5 also involved the pathway as death receptors and bind CD95 and TRAIL, respectively. All of the ligand binding to receptors will lead, with the help of the adapter proteins (FADD/ TRADD) to recruitment, dimerization, and activation of a caspase cascade and eventually cleavage of both cytoplasmic and nuclear substrates. To date, the best-characterized ligands and corresponding death receptors include CD95/Fas, TNF-α/TNFR1, Apo2L/DR4 and Apo2L/DR5.
Receptor trimerization results in recruitment of several death domains and eventually recruitment and activation of caspase-8 and caspase-10. Active caspase-8 and caspase-10 then either initiate apoptosis directly by cleaving and thereby activating executioner caspase-3/6/7), or activates the intrinsic apoptotic pathway through cleavage of the BID to induce efficient cell death. Immuno-blot analysis also revealed that the caspase-6 inhibitor blocked the cleavage of lamin A/C, whilst the caspase-3/7 inhibitor blocked the cleavage of poly (ADP-ribose) polymerase (PARP). Activation of caspase-8 may be prevented by FLICE inhibitory protein (FLIP).
Taken together, these results suggest that activation of caspases, the subsequent cleavage of lamin A/C and PARP, and the NFkB pathway are involved in the extrinsic pathway of cell apoptosis.