This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Interleukin-2 (IL-2) is a biological response modifier (cytokine) that promotes the growth, proliferation, and subsequent differentiation of disease-fighting blood cells such as T-Cells, B-Cells, NK (Natural Killer) cells, monocytes, macrophages, and oligodendrocytes. It is a potent immunoregulatory lymphokine originally referred to as "T-Cell growth factor" that is primarily secreted by Antigen-Activated T Cells (Ref.1). Human interleukin-2 (IL-2) is a 133-amino acid polypeptide with a molecular weight of 15-18 kDa. The Antigen-primed THC (T Helper Cell) secretes IL-2 in an autocrine manner, stimulating both itself and neighboring Antigen-primed T Cells to proliferate (T-Cell activation and proliferation).
T-cells require IL-2 as a growth factor for long-term culture, and this cytokine is critical for achieving T-cell-mediated immunity and thus for immune response regulation.
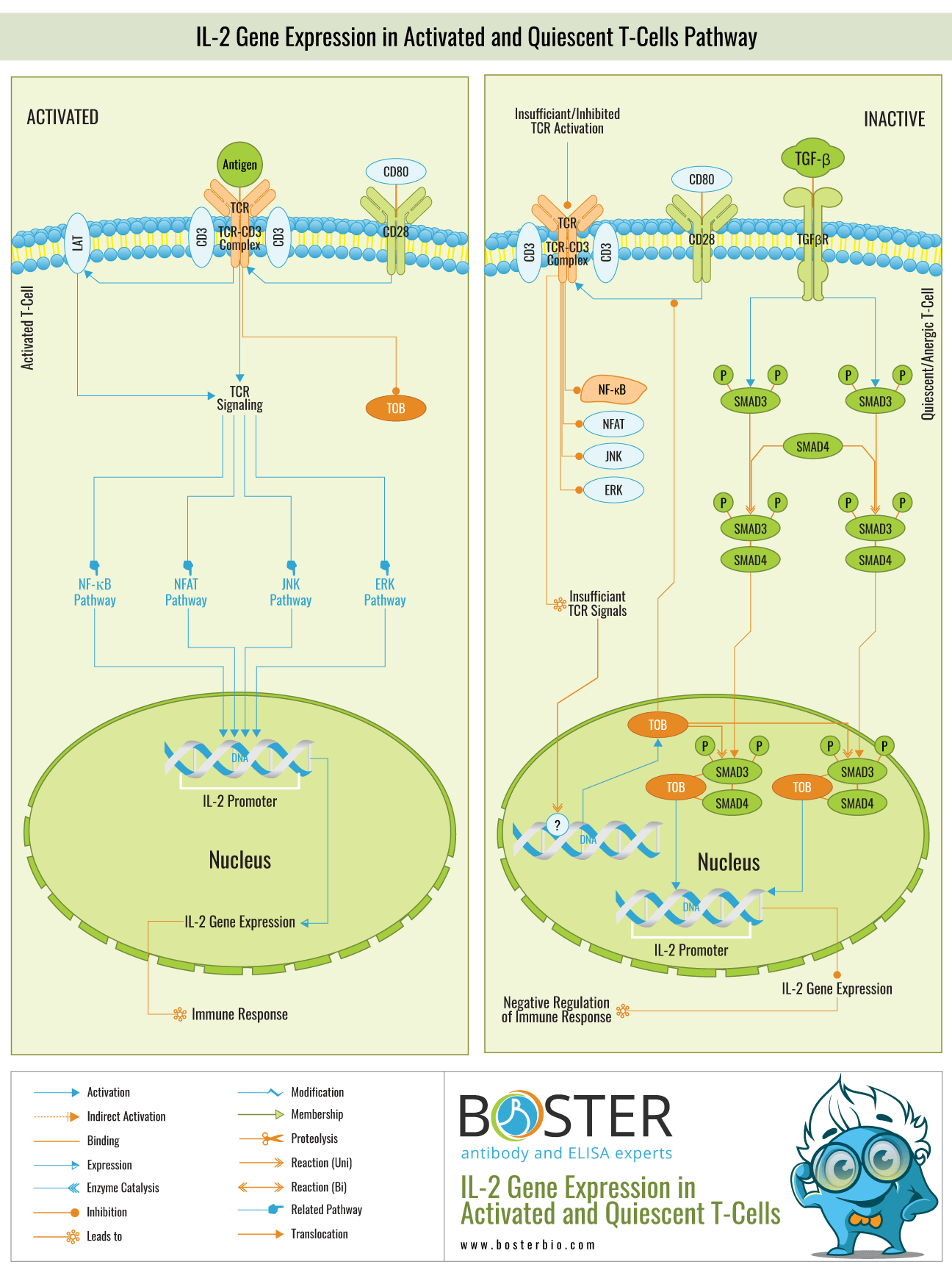
In normal T-Cells, engagement of TCR-CD3 complexes, costimulation by CD28, and recruitment of LAT (Linker of Activated T-Cells) initiates a membrane-proximal cascade of tyrosine phosphorylation events that ultimately results in the activation of multiple pathways, including ERK (Extracellular Signal Regulated Kinase), JNK (c-Jun N-terminal Kinase). Given the critical role of IL-2 gene expression in orchestrating immune responses, deregulation of T-Cell function, whether due to a defect or an excess, has dire consequences for the organism, including immunodeficiency and autoimmunity . If the T-Cell response is excessive, it will result in excessive IL-2 expression, which will result in hyperactivation of the immune system, which may result in autoimmune disorders or tissue injury. Thus, regulation of T-Cell activation and maintenance of T-Cells in a quiescent and unresponsive state is critical for the immune system to function properly. Suboptimal cross-linking of the TCR by antigen, which frequently occurs under physiological conditions, is insufficient to induce IL-2 gene expression and thus does not result in a productive immune response, but rather in T-Cell quiescence.
Unstimulated T-lymphocytes may be quiescent due to a lack of activation signals or the presence of inhibitory signals. Unlike primary unstimulated T cells, which can enter the cell cycle and clonally expand in response to antigen stimulation, anergic T cells do not proliferate. Rather than that, they persist in a state of long-term antigen-specific inactivity, referred to as T-Cell anergy. Both the quiescent and anergic states are caused by abnormal IL-2 gene expression.
The activation signals (Antigens) in quiescent T-Cells are either absent or insufficient to initiate a productive immune response. Additionally, the presence of inhibitory signals results in quiescence and anergy (Ref.2 & 4). The activation signals alone are insufficient to activate the multiple pathways involved in the activation of the IL-2 gene promoter region, including ERK, JNK, NF-KappaB, and NFAT. As a result, transcription of the IL-2 gene is inhibited. On the other hand, inhibitory substances inhibit the ERK, JNK, NF-KappaB, and NFAT pathways in both anergic and quiescent states. TOB (Transducer of ERBB2), a negative regulator of IL-2 transcription and T-cell proliferation, is expressed constitutively in unstimulated peripheral blood T lymphocytes and selectively in anergic T lymphocytes.
TOB is a member of the anti-proliferative TOB and BTG (B-Cell Translocation Gene) protein families (Ref.5). It is expressed at a maximum level in unstimulated and anergic T-Cells and at a minimum level in activated T-Cells. Once expressed, TOB inhibits CD28-mediated costimulation of TCR signaling, thereby repressing the pathways that normally result in the expression of IL-2. Normally, TOB expression is not observed in activated T cells. Otherwise, T-Cell activation requires down-regulation of TOB. The interaction of TOB with SMAD proteins results in the repression of the IL-2 gene. TOB enhances the inhibitory effect of SMAD proteins on TCR-CD3 and CD28–mediated IL-2 gene transcription. TOB increases SMAD binding to the IL-2 promoter's -105 negative regulatory element.
SMADs mediate TGF-Beta (Transforming Growth Factor-Beta)-induced signals in T cells via its receptors, TGF-BetaRII and TGF-BetaRI. The activated TGF-BetaRs phosphorylate the downstream targets SMAD2 and SMAD3, which colocalize with SMAD4 and translocate to the nucleus. The SMAD complex inhibits IL-2 gene expression by binding to the IL-2 promoter's -105 negative regulatory element.
TOB enhances the binding of SMADs to the IL-2 promoter's -105 negative regulatory element, which suppresses IL-2 expression. Because IL-2 plays a variety of critical roles in orchestrating immune responses, a thorough understanding of its expression and repression would enable us to better manipulate the immune system in order to overcome diseases such as cancer, infection, and autoimmune diseases. Not only are mature T lymphocytes stimulated to produce it, but certain T-Cell lymphoma cell lines also produce it constitutively. IL-2 is the primary tool in cytokine immunotherapy, which is used to prevent or delay disease progression by stimulating cell-mediated immunity (i.e., immune reconstitution) via exogenous T helper cell cytokines. IL-2 has been successfully used to treat a variety of human cancers (Metastatic Melanoma, Acute Myelogenous Leukemia, and Metastatic Renal Cell Carcinoma), and has recently been shown to improve immune responses in patients with HIV-1 (Human Immunodeficiency Virus Type-1) disease.