This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Nucleotide excision repair (NER) is the primary mechanism by which mammals remove large DNA lesions such as those caused by ultraviolet light, environmental mutagens, and some cancer chemotherapeutic adducts. NER deficiency is associated with the extremely common inherited skin cancer disorder xeroderma pigmentosum. Although the basic NER reaction and the factors that carry it out have been known for some time, recent studies have provided a much more detailed understanding of the NER mechanism, how it operates in the context of chromatin, and how it interacts with other cellular processes such as DNA damage signaling and transcription.
Nucleotide excision repair (NER) is the main pathway responsible for the removal of bulky DNA lesions induced by UV irradiation, environmental mutagens, and certain chemotherapeutic agents.
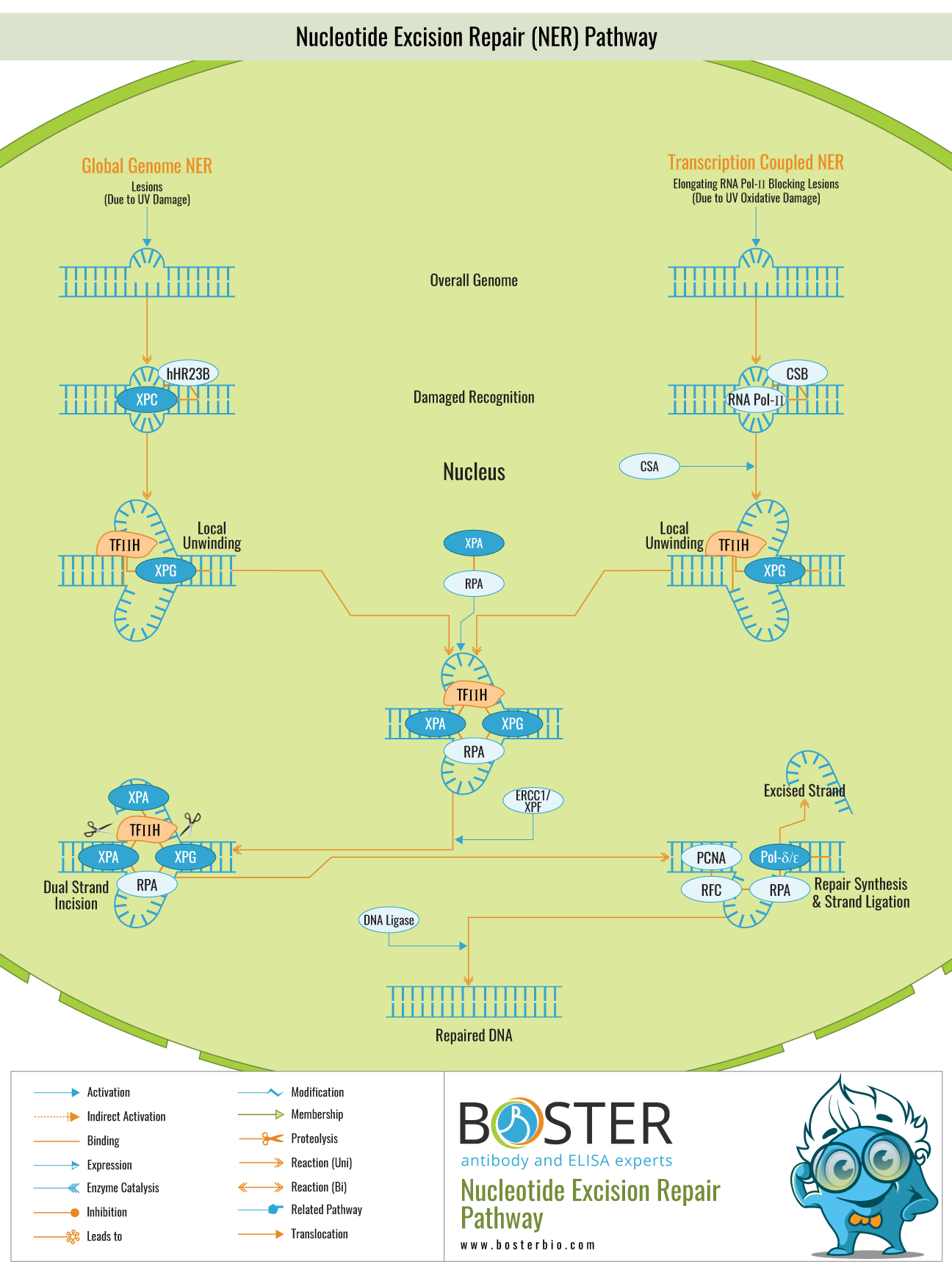
The molecular mechanism of NER is as described in the introduction in five steps. This step of damage recognition can be global or transcription-coupled. Global repair occurs throughout the genome and is transcription-independent. TCR occurs exclusively for damage to the transcribed strand of actively transcribed genes and is based solely on the stalling of an elongating RNA polymerase at the damage site. The initial studies to elucidate the molecular mechanism of NER were conducted in E. coli, followed by human studies. Thus, this section will begin by describing the molecular mechanism of NER in E. coli before moving on to the human mechanism.
There are two types of NER: repair of lesions throughout the genome, referred to as global genome NER (GG–NER), and repair of lesions in transcribed DNA strands, referred to as transcription-coupled NER (TC–NER). The majority of XP subgroups have defects in a component common to both NER subpathways. GG–NER requires the activity of all of the above-mentioned factors, as well as the GG–NER-specific complex XPC–hHR23B. The rate of GG–NER repair is highly dependent on the type of lesion. For example, 6-4PPs are removed from the genome much more rapidly than CPDs, most likely due to differences in the affinity of the damage sensor XPC–hHR23B. Additionally, the location (accessibility) of a lesion has an effect on the rate of removal in vivo. When the elongating RNA polymerase II complex comes into contact with a lesion in TC–NER, it detects damage. Interestingly, a distinct disorder known as Cockayne syndrome (CS) is associated with a specific transcription-coupled repair defect. The identification of two complementation groups (CS-A and CS-B) demonstrates that at least two distinct gene products are required for rapid and efficient transcribed strand repair. CS is a highly pleiotropic condition characterized by photosensitivity in addition to severe neurological, developmental, and premature aging characteristics. The majority of these symptoms are absent in completely NER-deficient XP patients. Additional CS symptoms suggest that transcription-coupled repair and/or the CS proteins serve a purpose other than NER.
Additionally, lesions that are not NER-specific (such as oxidative damage) but stall transcription elongation appear to be removed in a transcription-coupled manner, linking a blocked polymerase to multiple repair pathways. Surprisingly, some XP-B, XP-D, and XP-G patients exhibit CS characteristics in addition to XP manifestations. Several other XP-B and XP-D individuals have the brittle-hair syndrome trichothiodystrophy, which is similar to CS (TTD). This clinical conundrum suggests that these NER factors may have additional functions. A recent mouse model of TTD established a link between mutations in the XPD subunit of the dual functional TFIIH complex and deficiencies in basal transcription, which are thought to underpin at least some of the TTD manifestations. As a result of the additional functions of the NER factors involved, NER defects are associated with a surprising amount of clinical heterogeneity.