This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
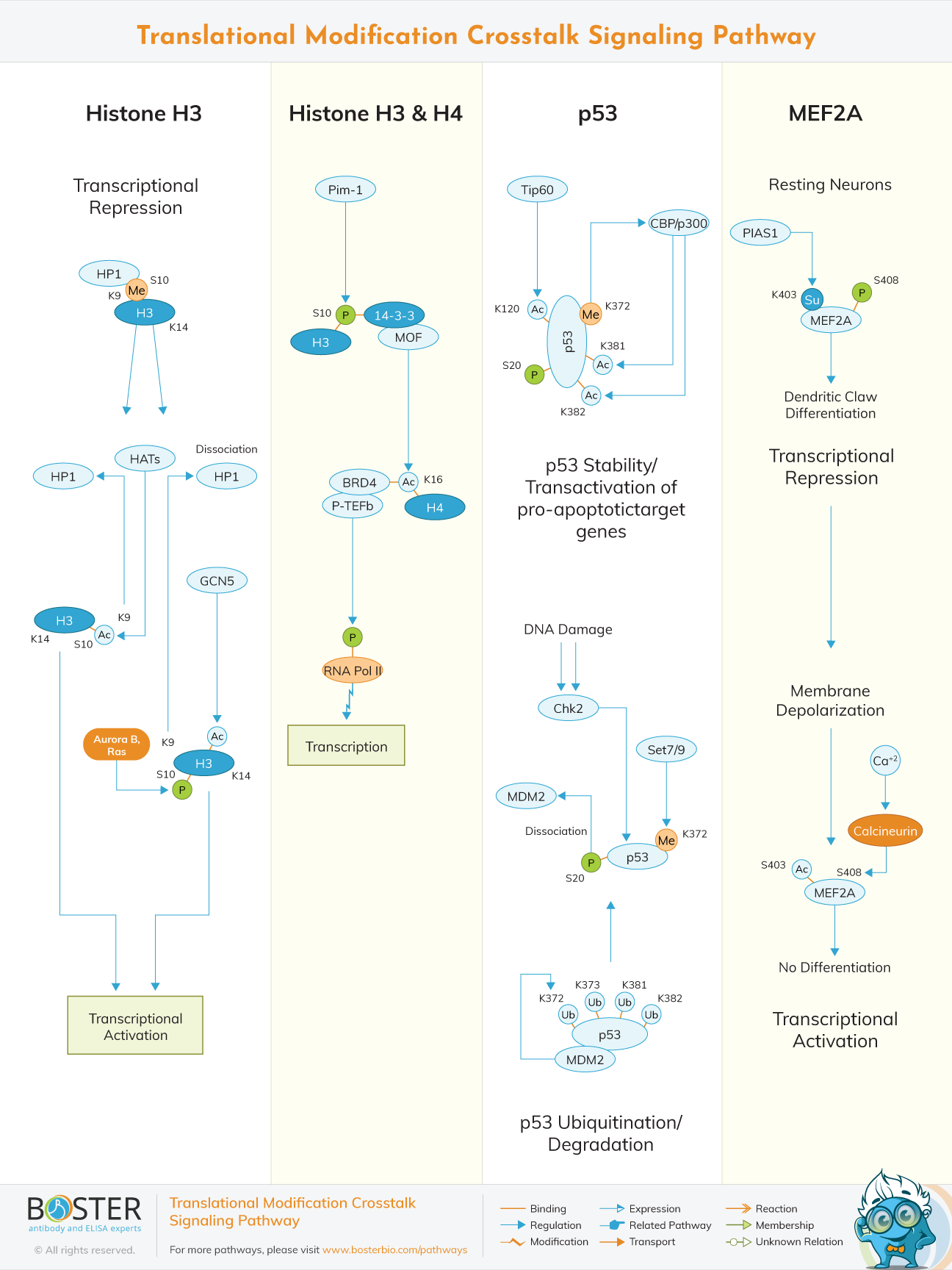
Post-translational modifications (PTMs) are emerging as major effectors of protein function, and in turn, cellular processes. Post-translational modifications include methylation, acetylation, phosphorylation and sumoylations. The interplay between PTMs is essential for proper gene expression, genome organization, cell division and DNA damage response. PTMs can directly impact cell function by modifying histones, modifying enzymes, assembling protein complexes as well as recognition and targeting in the genome or to other cellular compartments. An example of single modifications and gene expression is acetylation of certain lysines in Histone 3(H3) lysine 9th residue (K9) correlates with activation, while tri-methylation of this same residue is most often associated with compaction and gene repressiont. Most PTMs do not exist alone in the chromatin environment and the combination of these states can reinforce one another. For example, one PTM can serve as a docking site for a binding domain called a “reader” within one protein, while another “reader” within the same protein can recognize another residue.
This is the case for the reader protein BPTF, which binds both trimethylated 4th lysine residue on H3 (K4me3) and acetylated K16 of histone 4 (H4). Therefore, modulating the various types and degrees of modifications will impact output. For these reasons, the cell has developed a series of enzymes that are important for establishing and maintaining these PTMs, often called “writers” like histone methyltransferases, acetyltransferases, etc or “erasers” like histone demethylases, deacetylases, etc.
Methylated 9th lysine residue (k9) of H3 and the heterochromatin protein (HP1) that binds to the methyl residue causes transcription repression. Presence of HP1 means that the chromatin is a heterochromatin meaning that the chromatins is condensed inhibiting transcription. Transcription activation involving H3 is done in two ways. The first way involves the phosphorylation of 10th serine residue (S10) by Aurora B and Ras which mitotic kinases. Inhibition of these two kinases causes retention of HP1 proteins on mitotic chromosomes. The phosphorylation causes dissociation of HP1 and demethylation of k9.
The GCN5, a type of Histone acetyltransferase, acetylates k14 which causes the H3 to relax thus activating transcription. The second way involves acetylation of S10 by histone acetyltransferase (HATs) which leads the dissociation of HP1. This causes the H3 to relax encouraging transcription.
Serum stimulation induces the proto-oncogene serine/threonine protein Kinase (PIM-1) phosphorylates S10 of the preacetylated histone H3. The adaptor protein 14-3-3 binds the phosphorylated nucleosome and recruits the histone acetyltransferase MOF, which triggers the acetylation of histone H4 at lysine 16 (k16) residue. This histone crosstalk generates the nucleosomal recognition code determining a nucleosome platform for the bromodomain protein BRD4 binding.
The recruitment of the positive transcription elongation factor b (P-TEFb) via BRD4 induces the release of the promoter-proximal paused RNA polymerase II via phosphorylation which activate transcriptional elongation.
p53 is a transcription factor that suppresses tumor growth through regulation of dozens of target genes with diverse biological functions. The onco-protein Mdm2 is the primary E3 ubiquitin ligase for the p53 tumor suppressor.MDM2 negatively regulates p53 by targeting the ubiquitin ligase activity of MDM2. A complementary approach to prevent p53 degradation by MDM2 is to develop agents designed to inhibit the E3 ligase activity of MDM2. Ubiquitination of p53 increases p53-mediated transcriptional activation of genes involved with cell cycle arrest, but not apoptosis. MDM2 ligates ubiquitination of lysine residues k372, 373, 381and 382. p53 is thought to potentiate genomic stability, and consequently inhibit tumorigenesis, is by initiating cell cycle arrest, thus allowing repair of damaged DNA prior to DNA synthesis or segregation of the genome. Ubiquitination of p53 doesn't leave room for DNA repair.
CHK2, a serine-threonine kinase involved in DNA repair, cell cycle arrest or apoptosis in response to DNA damage, phosphorylate s20 residue of p53 causing the dissociation of MDM2. SET 7/9, a protein lysine methyltransferase, methylates k372 residue of p53.
Tip60 protein, which is a histone acetyltransferases, acetylates k120 residue of p53. Methylation of k372 causes p300/CBP, which has the ability to acetylate a wide variety of proteins, acetylates k381 and k382 residues. This reinforces p53 stability and transactivation of pro-apoptotic target genes.
Myocyte enhancer factor-2 (MEF2A) proteins are a family of transcription factors which through control of gene expression are important regulators of cellular differentiation and consequently play a critical role in embryonic development. MEF2A is a member of the myocyte enhancer factor-2 (MEF2) family of transcription factors. It's a transcription factor that participates in vascular development.In resting neurons, the s408 is phosphorylated and PIAS1, a member of the protein inhibitor of activated STAT (PIAS) family which possesses SUMO E3 ligase activity, sumoylates k403 residue of MEF2A. This activates dendritic claw differentiation which causes transcription repression.
During neuron membrane depolarization, Ca2+ activates calcineurin (CaN) which is a calcium and calmodulin dependent serine/threonine protein phosphatase. Calcineurin desumolyates s408 residue. The s403 is acetylates and this causes no differentiation which leads to transcription activation.