This website uses cookies to ensure you get the best experience on our website.
- Table of Contents

3 Citations 5 Q&As

1 Citations

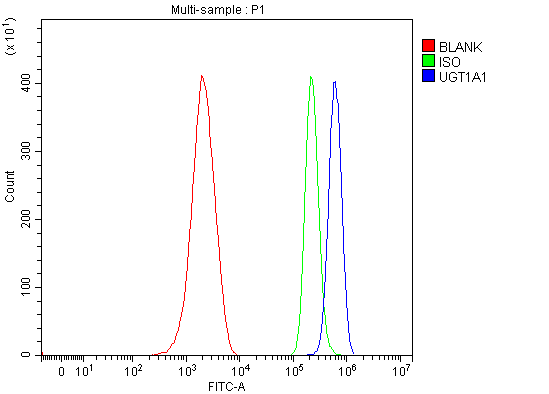
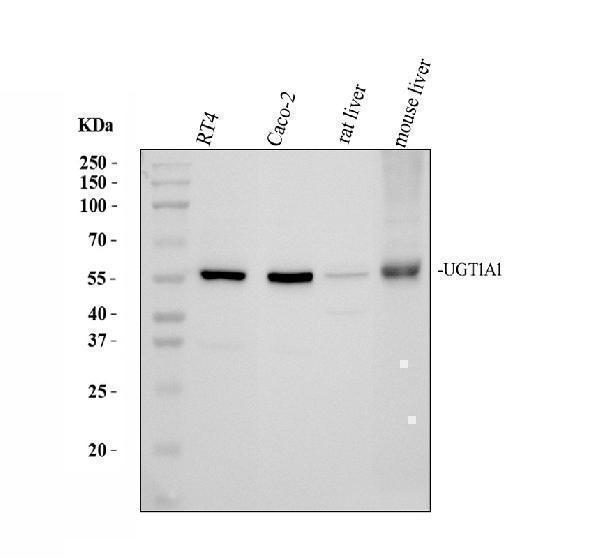
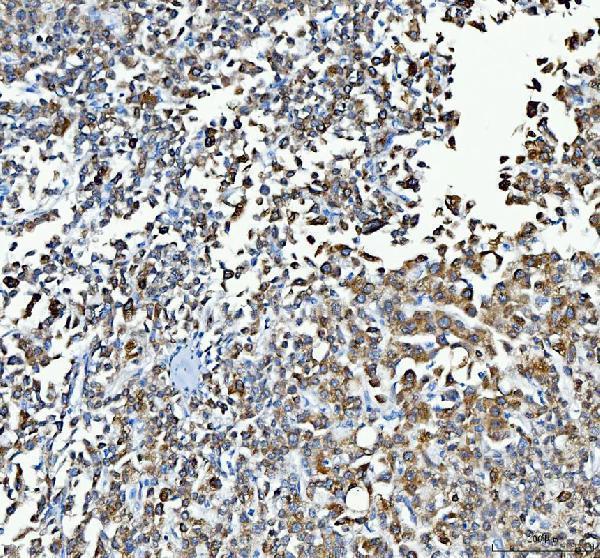
Facts about UDP-glucuronosyltransferase 1-1.
.
| Rat | |
|---|---|
| Gene Name: | Ugt1a1 |
| Uniprot: | Q64550 |
| Entrez: | 24861 |

| Belongs to: |
|---|
| UDP-glycosyltransferase family |

bilirubin UDP-glucuronosyltransferase 1-1; bilirubin UDP-glucuronosyltransferase isozyme 1; Bilirubin-specific UDPGT isozyme 1; GNT1; GNT1EC 2.4.1.17; HUG-BR1; UDP glucuronosyltransferase 1 family, polypeptide A1; UDP glycosyltransferase 1 family, polypeptide A1; UDP-glucuronosyltransferase 1-1; UDP-glucuronosyltransferase 1-A; UDP-glucuronosyltransferase 1A1; UDPGT 1-1; UDPGT; UG-BR1; UGT1; UGT1*1; UGT1.1; UGT1-01; UGT1A; UGT-1A; UGT1A1; UGT1AUDPGT 1-1; UGT1BILIQTL1
Mass (kDA):
59.663 kDA

| Rat | |
|---|---|
| Location: | 9q35 |
| Sequence: | 9; |

PMID: 7603447 by Coffman B.L., et al. Cloning and stable expression of a cDNA encoding a rat liver UDP- glucuronosyltransferase (UDP-glucuronosyltransferase 1.1) that catalyzes the glucuronidation of opioids and bilirubin.
PMID: 7608130 by Emi Y., et al. Drug-responsive and tissue-specific alternative expression of multiple first exons in rat UDP-glucuronosyltransferase family 1 (UGT1) gene complex.