This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
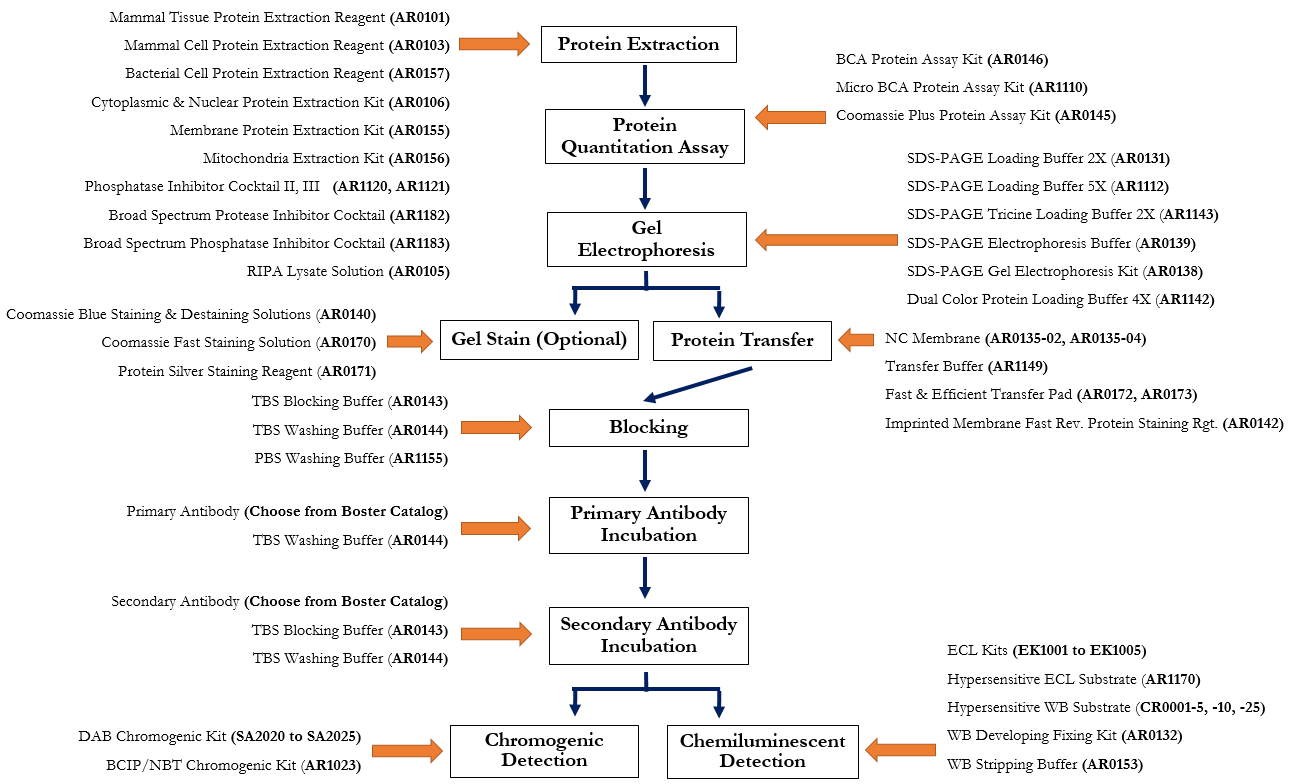
Western blotting, also known as immunoblotting, is a core method in molecular biology for detecting specific proteins within a complex sample. Developed in 1979 by W. Neal Burnette, Western blotting—also called immunoblotting—is a widely used method for detecting specific proteins in a sample. Adapted from earlier blotting techniques, it remains a standard in molecular biology. The process involves separating proteins by gel electrophoresis, transferring them to a membrane such as PVDF membranes or nitrocellulose, and using antibodies for detection.
Western blotting is commonly used to confirm protein expression and post-translational changes. It plays a key role in research and diagnostics, including HIV detection and studies of cancer and neurodegenerative diseases. Its reliability makes it essential in protein analysis workflows. Researchers often start with cell culture experiments and prepare extracts using cell lysis buffers containing protease inhibitor cocktail and phosphatase inhibitors to preserve protein integrity before analysis.
Let Bosterbio handle your Western blot CRO services and be your long term partner in broadening the bandwidth of your lab/team in a flexible manner.
The first step of gel preparation is to determine the gel percentage based on the molecular weight of your protein sample:
| Protein Size (kDa) | >100 | 30-100 | 10-30 | <10 |
|---|---|---|---|---|
| Gel Percentage | 8% | 10% | 12% | 15% |
If you are not sure of the size of your protein or are looking at proteins of a variety of molecular weights, then a gradient gel may provide the best resolution.
Notes
Dual Color Protein Loading Buffer is designed to prevent protein degradation during sample heating prior to electrophoresis and is able to work against pH changes during SDS-PAGE run. Many proteins are sensitive to pH changes that result from temperature fluctuations of Tris buffers during electrophoresis. It contains two tracking dyes: blue (Bromophenol Blue) for tracking the progress of electrophoresis and pink (Pyronin Y) for monitoring protein transfer to the membrane. Refer to the datasheet on our website for more information.
After electrophoresis, we recommend using one of our gel staining solutions to determine if the electrophoretic separation worked. Please refer to the datasheet(s) on our website for more information.
Note: Stained gel cannot be used in the subsequent protein transfer procedure.

We recommend using one of the filter papers from Boster:
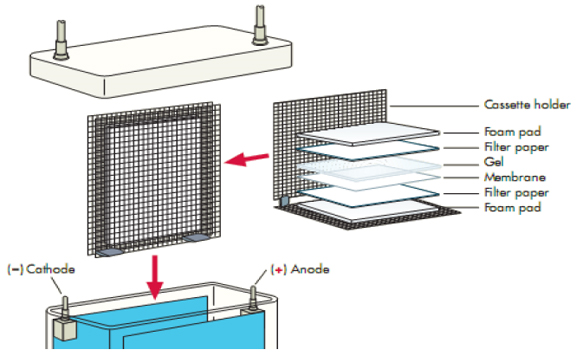
After transfer, membranes must be blocked to reduce background. Washing steps typically include buffers like Tris Buffered Saline or Phosphate Buffered Saline supplemented with Tween 20.
After blocking, the membrane is incubated with a primary antibody (that binds to the target protein) followed by an enzyme conjugated secondary antibody (HRP- or AP-conjugated secondary antibody).
Wash the membrane 3 times with the TBS Wash Buffer for 10 minutes each to remove the unbound antibodies.
Wash the membrane 3-6 times with the TBS Wash Buffer for 10 minutes each to remove unbound antibodies.
Signal detection can be carried out using chemiluminescent, colorimetric, or fluorescent methods. The choice depends on the label or enzyme conjugated to the secondary antibody. Common options include enhanced chemiluminescence (ECL) for high sensitivity and DAB chromogenic substrates for straightforward visualization. Fluorescent detection systems are also increasingly used, especially for multiplex analysis, and are compatible with PVDF membranes.
In this section, we provide the protocols for the Enhanced Chemiluminescence Detection (ECL) and colorimetric detection (DAB) methods. Use the method that fits your preferences and criteria.
Boster ECL chemiluminescent system depends on incubation of the western blot with a substrate that will luminesce when exposed to HRP on the secondary antibody. The light is then detected and captured by photographic film.
| Origin of Primary Antibody Species | Catalog # of ECL Kit* |
|---|---|
| Mouse IgG | EK1001 |
| Rabbit IgG | EK1002 |
| Goat IgG | EK1003 |
| Rat IgG | EK1004 |
| Mouse IgM | EK1005 |
* Each kit has sufficient reagents for 800 cm2 of membrane.
† Instead of using the ECL kit which provides 1) chromogenic reagents A and B (20X concentrated; 5 mL), 2) blocking buffer and 3) HRP-conjugated secondary antibody, one may use one of the following standalone chromogenic reagents A and B from Boster:
| Reagent A | Reagent B | ||||
|---|---|---|---|---|---|
| Product | Conc. | Volume | Conc. | Volume | Catalog # |
| ECL Western Blotting Substrate | 1X** | 100 mL | 1X** | 100 mL | AR1170 |
| Hypersensitive WB Chemiluminescent Substrate | 20X | 5 mL | 20X | 5 mL | CR0001-5 |
| Hypersensitive WB Chemiluminescent Substrate | 20X | 10 mL | 20X | 10 mL | CR0001-10 |
| Hypersensitive WB Chemiluminescent Substrate | 20X | 25 mL | 20X | 25 mL | CR0001-25 |
** Ready-to-use
Record the test result using autoradiography film or CHEMIDOC in a darkroom. For autoradiography film, exposure time for the specific antigen depends on the developing effect, which could range from seconds to minutes.
Using WB Developing Fixing Kit (Boster Catalog # AR0132), develop and fix the film in a dark room immediately. Alternatively, fluorescence CCD scan, digital imager, or luminometer can be used.
Notes
Prepare DAB or BCIP/NBT substrate solution described below.
| Origin of Primary Antibody Species | Color | Catalog # of DAB Kit |
|---|---|---|
| Mouse IgG | Yellow | SA2020 |
| Goat IgG | Yellow | SA2021 |
| Rabbit IgG | Yellow | SA2022 |
| Rat IgG | Yellow | SA2023 |
| Mouse IgG | Blue | SA2024 |
| Rabbit IgG | Blue | SA2025 |
Learn the principle behind Western blotting, including protein separation by SDS-PAGE and target detection through membrane blotting and antibody-based analysis.
Review Western blot sample preparation guidance to help improve protein extraction, handling, and sample quality across different sample types.
Troubleshoot common Western blot issues such as weak signal, incorrect band size, smiling gels, and high background with practical tips.
Explore Western blot optimization tips for transfer, blocking, antibody incubation, and detection to improve assay performance and consistency.
Before you move on to blocking, make sure the membrane is worth taking forward. A Western blot can start failing long before the antibodies ever touch the membrane.
PVDF or Nitrocellulose? A practical guide to choosing the right membrane based on workflow, reprobing needs, and downstream use.
How to Strip and Re-probe Cleanly? A verification-first workflow to prevent ghost bands and high background.
Learn how to quantify Western blot results more reliably by understanding saturation, normalization, and linear-range signal measurement.