This website uses cookies to ensure you get the best experience on our website.
- Table of Contents

3 Citations
Facts about Alpha-ketoglutarate-dependent dioxygenase FTO.
Especially demethylates N(6)-methyladenosine (m6A) RNA, the most prevalent internal modification of messenger RNA (mRNA) in higher eukaryotes. Has no activity towards 1- methylguanine.
| Mouse | |
|---|---|
| Gene Name: | Fto |
| Uniprot: | Q8BGW1 |
| Entrez: | 26383 |

| Belongs to: |
|---|
| fto family |

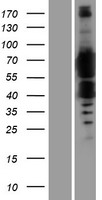
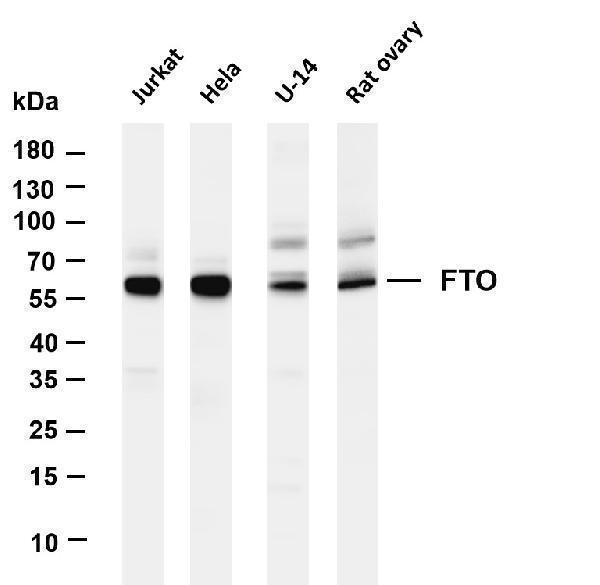
Alpha-ketoglutarate-dependent dioxygenase FTO; EC 1.14.11.-; fat mass and obesity associated; Fat mass and obesity-associated protein; FTO; KIAA1752protein fto; MGC5149
Mass (kDA):
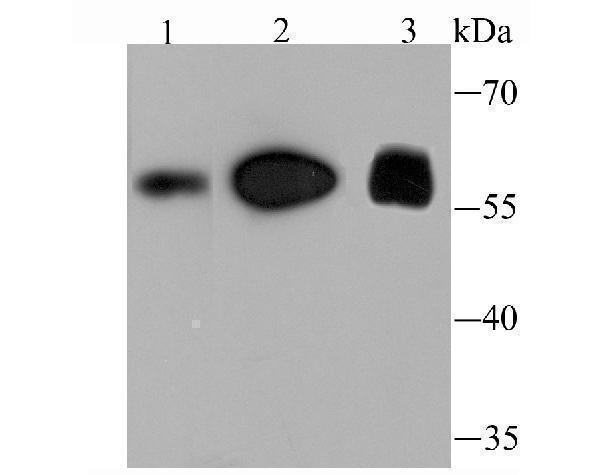
58.007 kDA

| Mouse | |
|---|---|
| Location: | 8 C4-C5|8 44.34 cM |
| Sequence: | 8; |
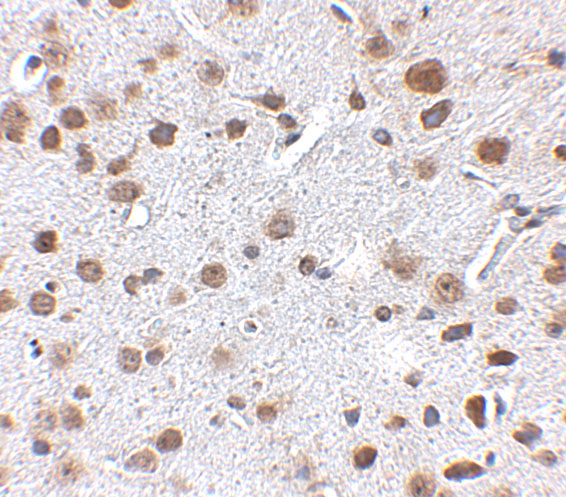
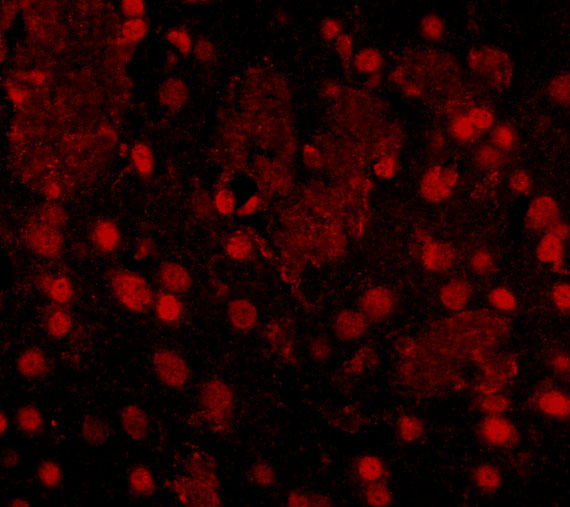
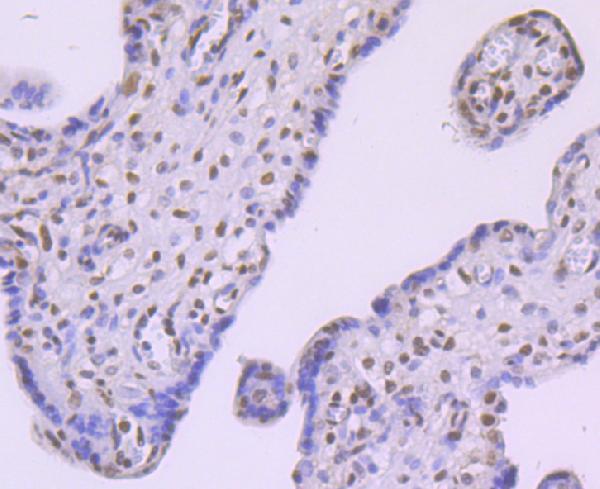
Ubiquitous. Detected in brain, brain cortex, hypothalamus, cerebellum, liver, pancreas, heart, kidney, white adipose tissue and skeletal muscle. Most abundant in the brain, particularly in hypothalamic nuclei governing energy balance.


PMID: 10501967 by Peters T., et al. Cloning of Fatso (Fto), a novel gene deleted by the Fused toes (Ft) mouse mutation.
PMID: 17991826 by Gerken T., et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase.
*More publications can be found for each product on its corresponding product page