This website uses cookies to ensure you get the best experience on our website.
- Table of Contents

1 Citations 4 Q&As
Facts about Very long-chain specific acyl-CoA dehydrogenase, mitochondrial.

.
| Human | |
|---|---|
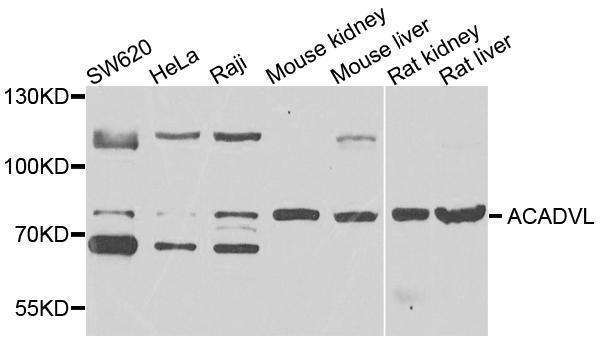
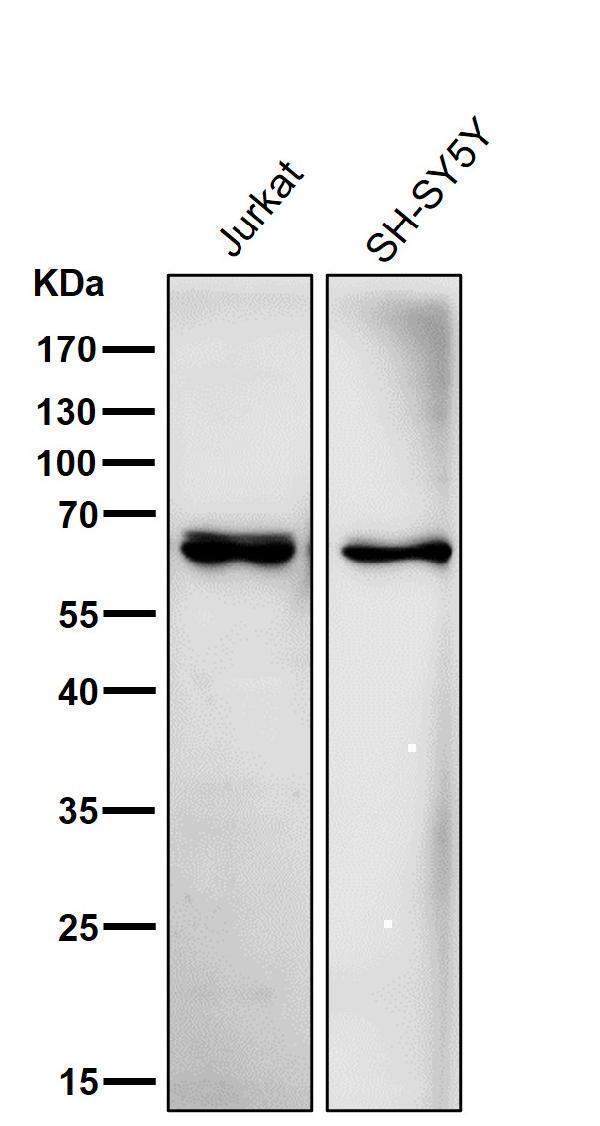
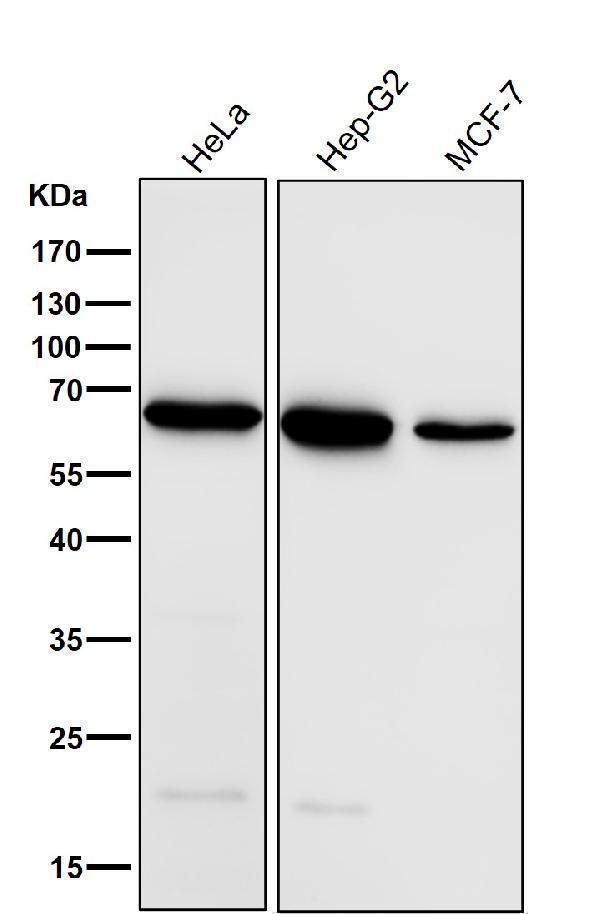
| Gene Name: | ACADVL |
| Uniprot: | P49748 |
| Entrez: | 37 |

| Belongs to: |
|---|
| acyl-CoA dehydrogenase family |

ACAD6; acyl-CoA dehydrogenase, very long chain; acyl-Coenzyme A dehydrogenase, very long chain; EC 1.3.99; EC 1.3.99.-; LCACD; very long-chain specific acyl-CoA dehydrogenase, mitochondrial; VLCAD
Mass (kDA):
70.39 kDA

| Human | |
|---|---|
| Location: | 17p13.1 |
| Sequence: | 17; NC_000017.11 (7217125..7225267) |
Mitochondrion inner membrane.


PMID: 7668252 by Aoyama T., et al. Cloning of human very-long-chain acyl-coenzyme A dehydrogenase and molecular characterization of its deficiency in two patients.
PMID: 8845838 by Andresen B.S., et al. Cloning and characterization of human very-long-chain acyl-CoA dehydrogenase cDNA, chromosomal assignment of the gene and identification in four patients of nine different mutations within the VLCAD gene.