This website uses cookies to ensure you get the best experience on our website.
- Table of Contents

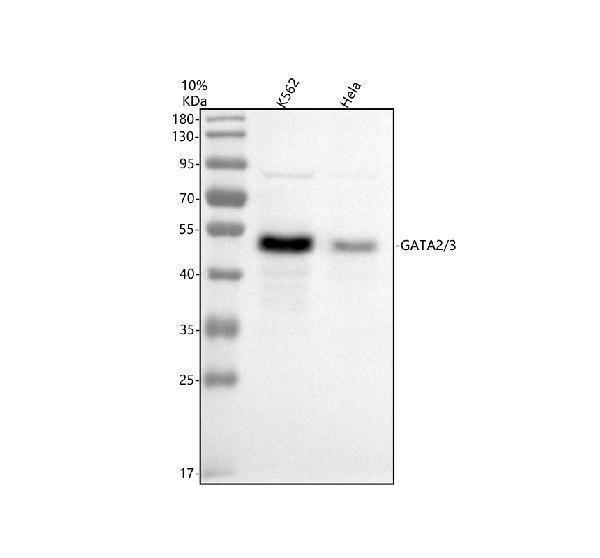
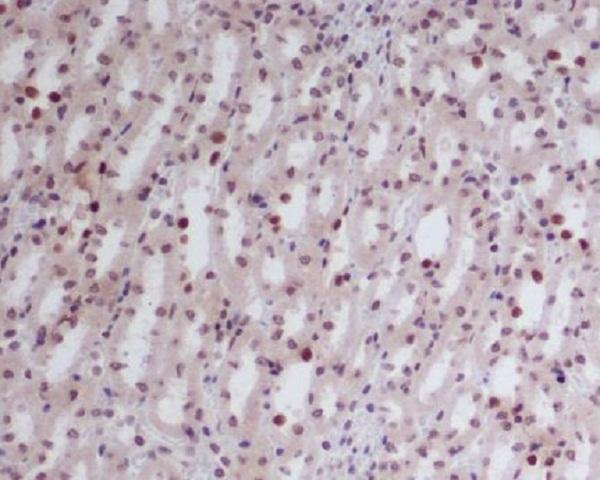
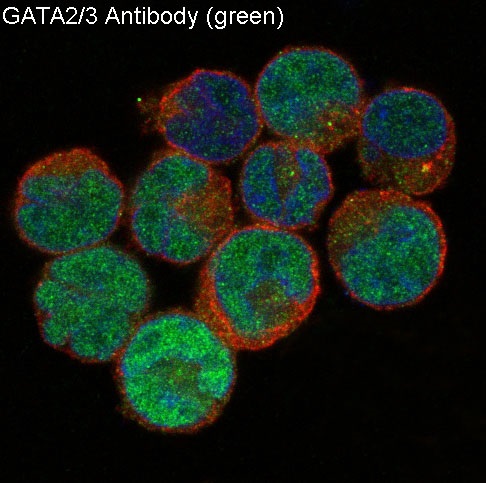
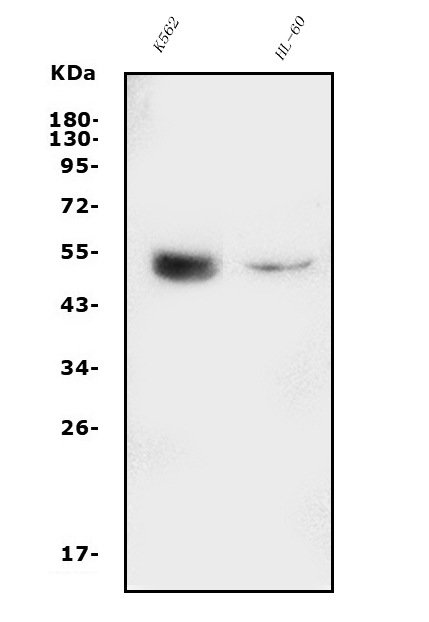
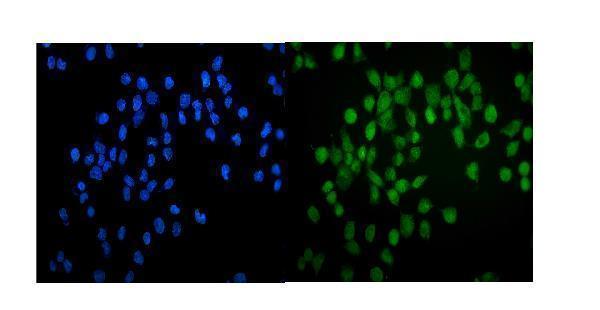
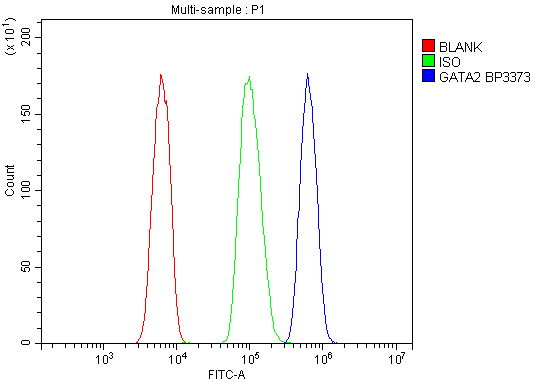
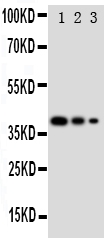
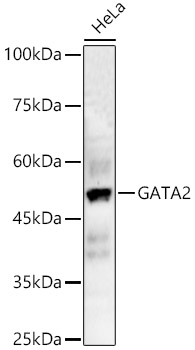
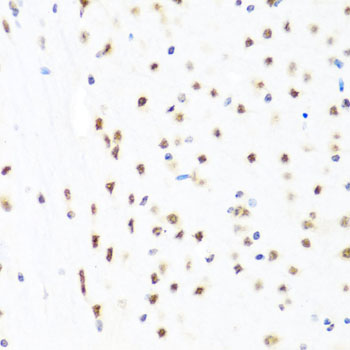
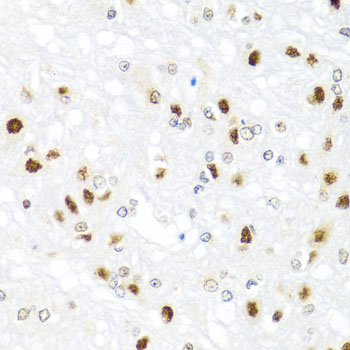
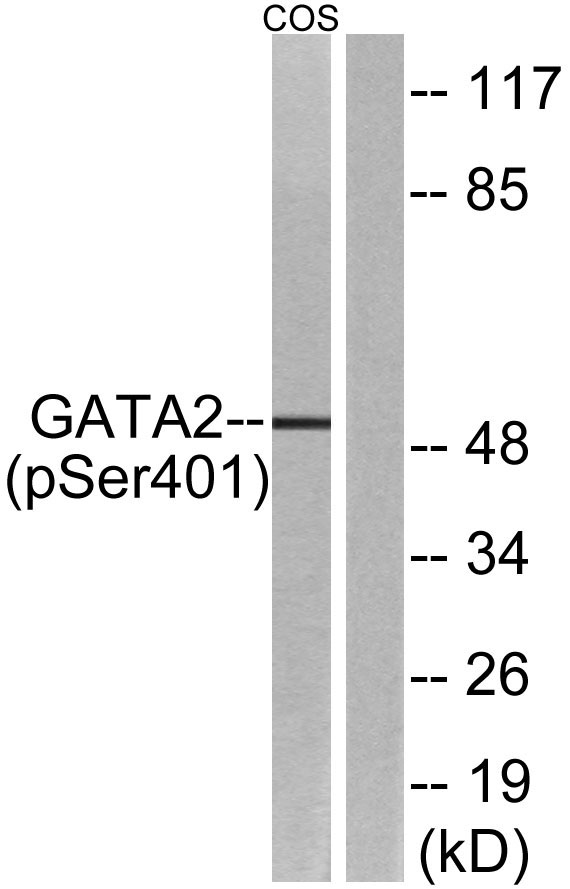
Facts about Endothelial transcription factor GATA-2.

.
| Human | |
|---|---|
| Gene Name: | GATA2 |
| Uniprot: | P23769 |
| Entrez: | 2624 |

| Belongs to: |
|---|
| No superfamily |

endothelial transcription factor GATA-2; FLJ45948; GATA binding protein 2; GATA2; GATA-2; GATA-binding protein 2NFE1B; MGC2306
Mass (kDA):
50.5 kDA

| Human | |
|---|---|
| Location: | 3q21.3 |
| Sequence: | 3; NC_000003.12 (128479422..128493201, complement) |
Endothelial cells.
Nucleus.


PMID: 1714909 by Lee M.-E., et al. Cloning of the GATA-binding protein that regulates endothelin-1 gene expression in endothelial cells.
PMID: 1370462 by Dorfman D.M., et al. Human transcription factor GATA-2. Evidence for regulation of preproendothelin-1 gene expression in endothelial cells.