This website uses cookies to ensure you get the best experience on our website.
- Table of Contents

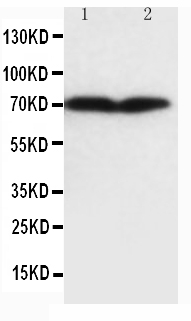
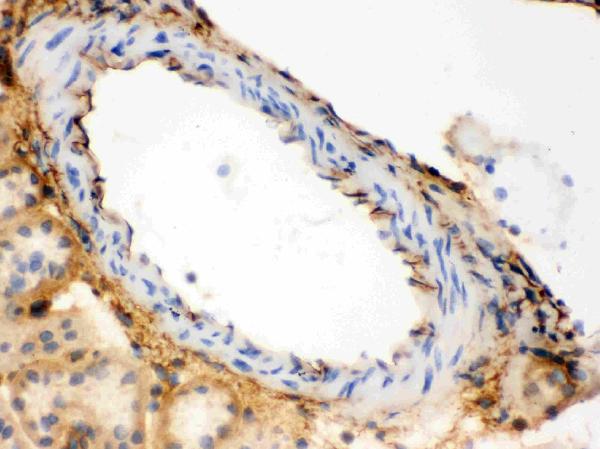
2 Citations 15 Q&As


43 Citations 16 Q&As
13 Citations 15 Q&As
6 Citations
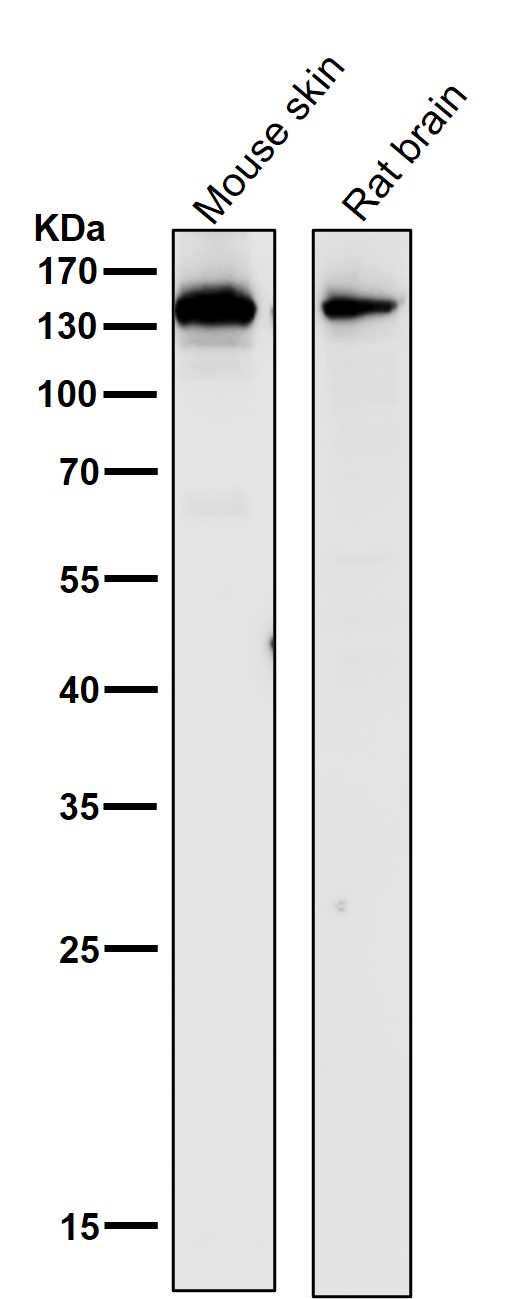
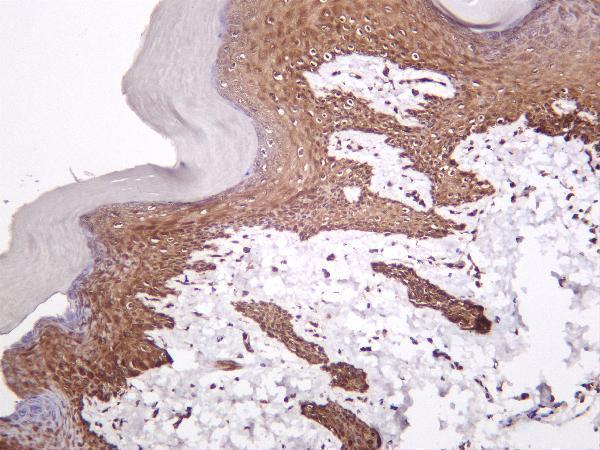
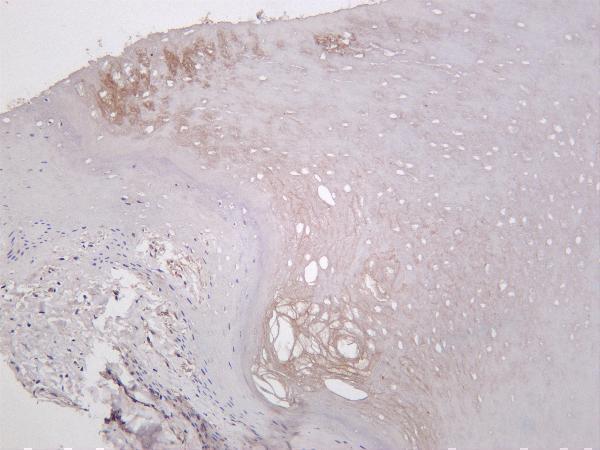
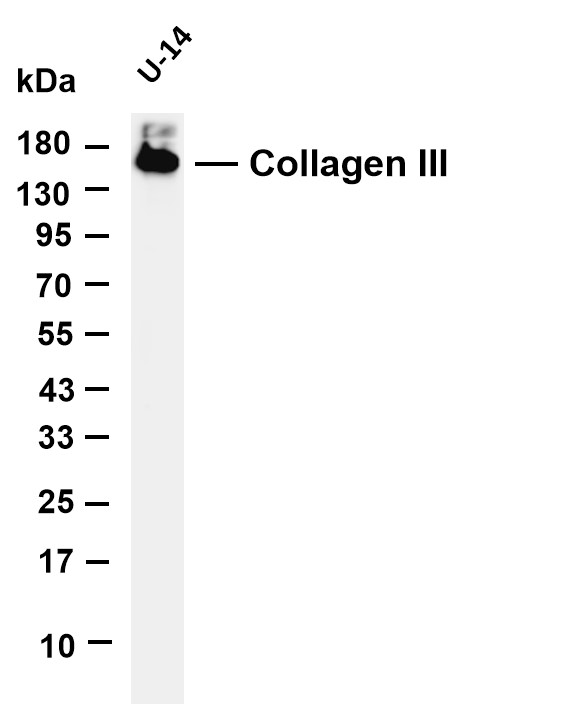
Facts about Collagen alpha-1(III) chain.

Is the significant ligand of ADGRG1 in the developing brain and binding to ADGRG1 inhibits neuronal migration and activates the RhoA pathway by coupling ADGRG1 to GNA13 and possibly GNA12. .
| Human | |
|---|---|
| Gene Name: | COL3A1 |
| Uniprot: | P02461 |
| Entrez: | 1281 |

| Belongs to: |
|---|
| fibrillar collagen family |

alpha1 (III) collagen; COL3A1; Collagen 3; collagen alpha-1(III) chain; Collagen III alpha 1; collagen, fetal; collagen, type III, alpha 1; Collagen-3; EDS4A; Ehlers-Danlos syndrome type IV, autosomal dominant; FLJ34534
Mass (kDA):
138.564 kDA

| Human | |
|---|---|
| Location: | 2q32.2 |
| Sequence: | 2; NC_000002.12 (188974373..189012746) |
Secreted, extracellular space, extracellular matrix.



PMID: 2764886 by Ala-Kokko L., et al. Structure of cDNA clones coding for the entire prepro alpha 1 (III) chain of human type III procollagen. Differences in protein structure from type I procollagen and conservation of codon preferences.
PMID: 11566270 by Valkkila M., et al. Genomic organization of the human COL3A1 and COL5A2 genes: COL5A2 has evolved differently than the other minor fibrillar collagen genes.
*Showing only the more recent 20. More publications can be found for each product on its corresponding product page