This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Facts about Menin.

Binds to the TERT promoter and represses telomerase expression. Plays a role in TGFB1-mediated inhibition of cell-proliferation, possibly regulating SMAD3 transcriptional activity.
| Human | |
|---|---|
| Gene Name: | MEN1 |
| Uniprot: | O00255 |
| Entrez: | 4221 |

| Belongs to: |
|---|
| No superfamily |

MEAI; MEN1; Menin; multiple endocrine neoplasia I; multiple
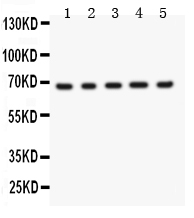
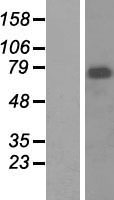
Mass (kDA):
68.023 kDA

| Human | |
|---|---|
| Location: | 11q13.1 |
| Sequence: | 11; NC_000011.10 (64803514..64811294, complement) |
Ubiquitous.
Nucleus. Concentrated in nuclear body-like structures. Relocates to the nuclear matrix upon gamma irradiation.



MEN1 is an autosomal dominant hereditary disorder. When it is mutated, it can cause germline mutations. Consequently, mutational analysis should be conducted by a laboratory for clinical genetics. Therefore it is recommended for patients suffering from pancreatic NETs to undergo MEN1 mutational analysis. This article will provide a brief overview of some of the most well-known applications of MEN1 mutant analysis.
The MEN1 gene is inherited as an autosomal dominant condition. The MEN1 gene mutation allows early detection in index patients and in affected family members by genetic tests. Early diagnosis is essential for the development of an appropriate diagnosis program, and preventing the cancer from progressing to the more severe stages of the disease including metastases and malignant progression. MEN1 is one of the most common causes of mortality and morbidity in patients affected by the hormone-overproduction syndrome. Understanding the causes of MEN1 tumorigenesis is vital in the development of new, targeted treatments.
Pituitary adenomas are found in anywhere from 15 to 90% of cases. The symptoms of this autosomal dominant genetic disease depend on the amount of hormones released by the pituitary gland and on the extent to which the tumor is causing compression of nerve tissue around it. In addition, there could be massive effects on the brain, including blurred vision or headache. Most pituitary tumours afflicted by MEN1 secrete prolactin, growth hormone or adenocorticotrophin. However, some are not. Treatment and detection of MEN1 related pituitary tumors usually require radiotherapy or surgery.
About 40% of those suffering from MEN1 will develop cancers in the digestive tract. Small tumors can develop at the same time. Some of them produce hormones and others will not, and some will be cancerous. The gastrinoma produces a hormone called gastrin. This hormone regulates stomach acid release. Gastrin overdoses can trigger Zollinger-Ellison syndrome. In addition to pancreatic cancer, patients with MEN1 can develop different kinds of gastrinomas on their bodies.
The symptoms of MEN1 can be life-threatening. Certain patients may develop Cushing's or acromegaly. X-ray CT scans can reveal such tumours. The majority of MEN1 patients have benign tumors. While the tumors might not cause any symptoms however, they can pose a serious health risk and can cause life-threatening complications.
The human MEN1 gene is located at 11q13. Patients who have mutations in this gene are at high chance of developing cancer. Patients with this variant of the gene suffer from mild disease and a longer life expectancy than the typical Dutch citizen. Mutation-negative MEN1 patients are more likely to have pHPT and PIT combination mutations. In addition, mutation-negative patients are considered index cases. This means they have no family history, with the exception that the patient's sibling has developed clinical MEN1 disease.
MEN1 mutations are typically caused by an anomaly in CDKN1B. This gene is vital for cell division and growth control. MEN1 mutations can be found in the germline in only a small proportion of people. The germline mutations affect the gene responsible for the cyclin dependent kinase inhibitors which regulate cell development and division. While there aren't any cures for MEN1-related disorders the research continues to discover more information about the condition and the root causes of it.
While there is currently no cure for MEN1 tumors however, there is a way to lower the chance of metastatic neuroendocrine tumors through the right genetic treatment. The MEN1 gene can be found on the long arm (11q13) of chromosome 11. It contains 10 exons that encode for the amino acid protein 610. Numerous MEN1 germline mutations have been discovered, that reduce or eliminate menin function.
Recent research has revealed a new MEN1 mutation. It involved an adenine insertion in codon 502, leading to frame shift and an abrupt stop codon. The mutated allele was homozygous for blood cells, hemizygous for two pancreatic tumors and homozygous for duodenal cancer. Immunohistochemical staining confirmed that the MEN1 c.1523G variant was associated with the growth of duodenal tumors.
Patients with insulinoma must undergo MEN1 mutational testing. Currently, the ENETS consensus guidelines recommend MEN1 mutational analysis for patients with insulinoma younger than 20 years. De Laat et al. suggested that mutational analyses should be expanded to include GEP-NET diagnosed before the age of 30. These findings will only be confirmed by larger groups of patients.
Since 2001 Since 2001, the MEN1 genetic test is available in a national center for referral. The study assessed 189 patients to determine germline mutations in MEN1 and analyzed 134 unrelated probands and 55 family members of patients with a mutation-positive pedigree. The study included people from all over Hungary between 2001 and 2017. Of these, 104 had a mutation which met Endocrine Society criteria in 2012. Due to the lack of family histories this study was unable to determine the path of the disease for all patients. The endocrinologist responsible for the study provided details on the clinical condition of patients.
If a MEN1 patient is identified as having a MEN1 tumor on imaging It is crucial to confirm the diagnosis on subsequent examinations. This approach is relatively new to the field of MEN1 research. The study also assessed MEN1 manifestations in time to identify possible mutations. Notably, there was only one positive mutation in this cohort of nationals. This is a clear indication of the rarity of these mutations however it also highlights the need for clinical genetics laboratories that can provide accurate results.
To be able to accurately diagnose MEN1, a patient has to be accompanied by two family members who have been clinically affected, and two generations of family members affected. A successful MEN1 family analysis will pinpoint the risk haplotype 11q13. But, this method can't be applied to a single index case. Therefore, reconstruction of haplotypes within families will allow for an more precise assessment of the inheritance of the disease.
All patients suffering from pancreatic NETs should undergo MEN1 mutational analysis. MEN1 mutational testing was not previously recommended. This was because the patient with the problem had an ancestral background. This technique is now more commonly used for the management of pancreatic NTEs. MEN1 mutational analysis was carried out in the study of 776 patients suffering from pancreatic cancer NETs.
The MEN1 gene is responsible for a high proportion of pancreatic NETs. In reality, 1 out of 3 pancreatic NETs is cancerous. Metastases are very common. A small percentage of metastases could spread to the lung or the liver. Patients with pancreatic NETs were recommended to undergo MEN1 mutational tests in the current study.
11q13 is the site of the MEN1 gene. The mutational analysis can be used to determine if family members are at risk for developing the disease. The results can also aid doctors in identifying relatives that are affected by the mutation, and also reduce the likelihood of cancer spreading. These results can be used to identify the MEN1 mutation in family members, if there is one.
The MEN1 mutational test confirms the clinical diagnosis. It also helps determine the relatives suffering from asymptomatic symptoms who should be screened early. The reality is that half of family members do not have the mutation and therefore should not be screened. This can relieve families of the burden of a affected family member. MEN1 mutational analysis is also able to resolve diagnostic problems due to phenocopies. These are found to affect 5% to 10% of families.
MEN1 is an inherited disorder which affects the pituitary and pancreas. In addition to the parathyroid gland the condition can also cause hypercalcemia. While the disease usually presents with long-term asymptomatic course, it could also result in hypercalcemia. Patients with MEN1 must have regular calciuria, imaging the urinary tract, as well in bone mineral density tests using dual-energyX-ray absorptiometry. Patients with MEN1 frequently develop multiple parathyroid adenomas over their life. The disease is correlated with a loss in heterozygosity at 11q13. This is a crucial step in the process of developing adenomas.
However the timing of genetic screening is critical. It isn't feasible to test all patients suffering from parathyroid tumors if there is no known instances of index cases. Genetic testing can be costly and time-consuming. Moreover, patients who carry MEN1 mutations should undergo regular screening for parathyroid tumours, regardless of age. This screening protocol is based on guidelines that have been published and a thorough review of literature.
All patients who have patients who have a MEN1 gene mutation should have their DNA tested. MEN1 mutations are rare and could represent about 2% of all cases. Genetic screening is recommended to determine if a patient carrying the MEN1 variant. There are numerous other reasons why MEN1 is recommended for patients with parathyroid tumors.
Although MEN1 does not necessarily mean you will be diagnosed, it may often confirm the diagnosis. The tumor's size and location are key indicators of malignancy. Patients with MEN1 tumors may be more likely to die than the general population. Although it isn't easy to determine if a cancer is cancerous, this test could be used to confirm the diagnosis.
PMID: 9103196 by Chandrasekharappa S.C., et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1.
PMID: 17555499 by Toledo R.A., et al. Novel MEN1 germline mutations in Brazilian families with multiple endocrine neoplasia type 1.