This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
During the 1960s, researchers discovered that the cell could destroy its own contents by wrapping it in membranes, generating sack-like vesicles that were carried to a recycling compartment called the lysosome for breakdown.Difficulties in investigating the process meant that nothing was understood until Yoshinori Ohsumi utilized baker's yeast to uncover genes needed for autophagy in a series of remarkable studies in the early 1990s. He then went on to clarify the fundamental principles of autophagy in yeast and demonstrated that mammalian cells utilise comparable complex machinery.
Ohsumi's discovery ushered in a new paradigm in our knowledge of how cells recycle their contents. His studies paved the way for a better understanding of the essential role of autophagy in numerous physiological processes, including as starvation adaptation and infection response. Autophagy gene mutations can cause illness, and the autophagic mechanism is implicated in a variety of disorders.
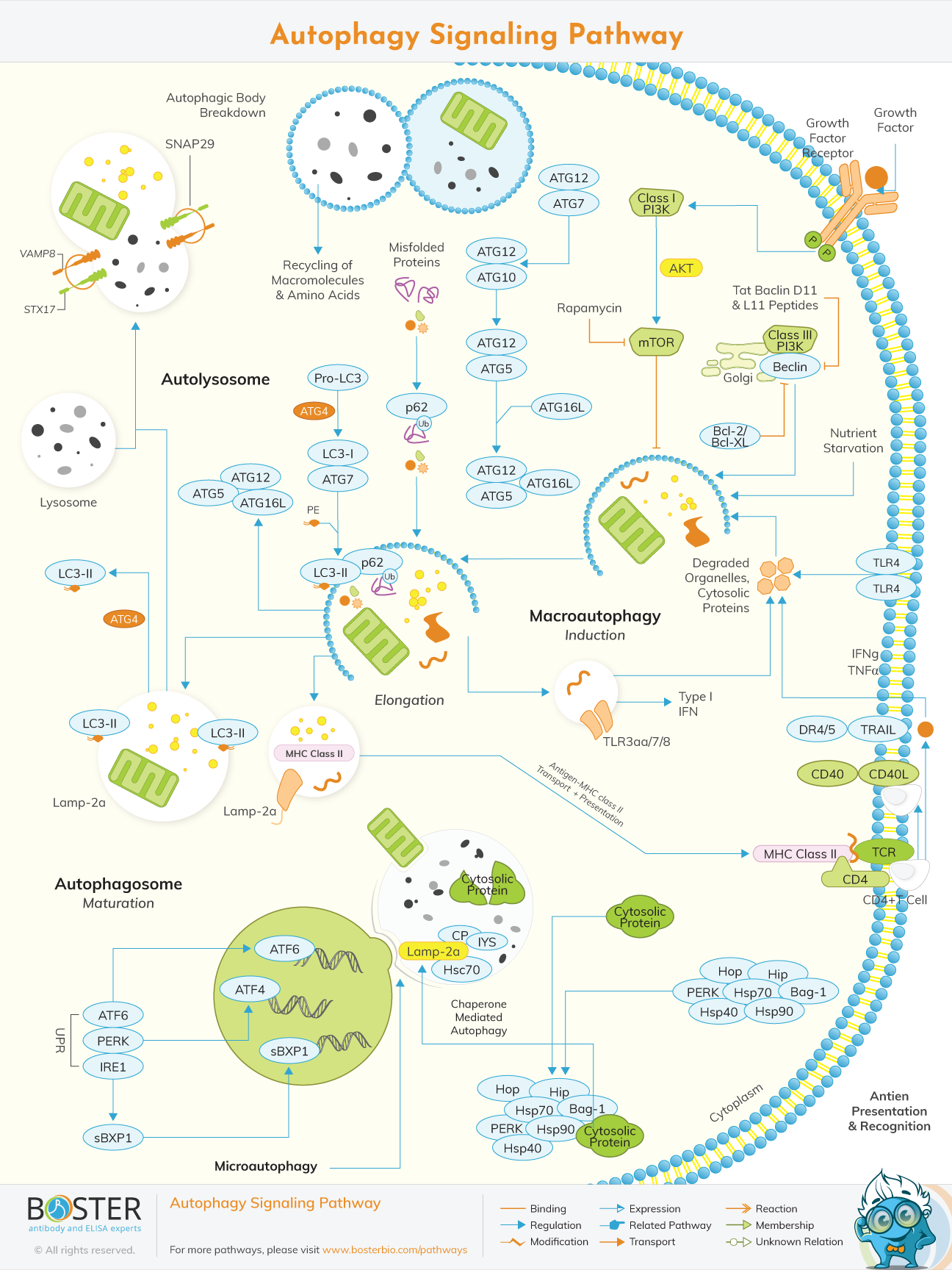
Autophagy is a cellular self-degradation mechanism in which double-membrane autophagosomes sequester organelles or portions of cytosol and fuse with lysosomes or vacuoles for hydrolase breakdown. Extracellular and intracellular stress and signs such as malnutrition, growth factor deprivation, ER stress, and pathogen infection increase autophagy.
Autophagy is characterized by the development of a double-membrane vesicle that encases cytoplasm, malformed proteins, long-lived proteins, and organelles before fusing with lysosomes for degradation. The double-membrane vesicle is formed through a complex process involving 16 autophagy-related proteins (Atg proteins). Apart from that, autophagy involves two ubiquitin-like conjugation processes. These systems generate modified autophagy regulator complexes, such as Atg8-PE and Atg5-Atg12-Atg16, which can influence autophagosome formation and size.The formation of the autophagosome is then nucleated, expanded, uncoated, and completed, enabling it to fuse with lysosomes.
Several conserved Atg (autophagy-related) proteins, the majority of which were first discovered in yeast, are involved in the molecular mechanism of autophagy. Two complexes are required for autophagosome formation to begin. Vps34, Atg6/Beclin1, Atg14, and Vps15/p150.73 are all class III PI3 Ks found in this complex. The Atg1 serine/threonine kinase is part of the other complex. Atg1's kinase activity is dependent on the activity of two other autophagy proteins, Atg13 or Atg8, and Atg17. Atg1 has been shown to interact with the Atg8 orthologues LC3 (microtubule-associated protein light chain 3), GATE-16 (Golgi-associated ATPase enhancer of 16 KDa), and GABARAP in mammals that lack Atg13 (G-amino butyric acid type A receptor-associated protein).
Autophagy is triggered in response to a variety of stressors and physiological situations. Food deprivation, heat, and hypoxia, for example, are all known environmental modulators of aging that can activate autophagy [8–10]. At the molecular level, an autophagy pathway has extraordinary interrelationships with variables that influence aging.
The IGF-1/Insulin signaling system, which is nutrient-responsive, promotes reproductive development, morphogenesis, and survival. The insulin/IGF-1 signaling pathway in C. elegans suppresses dauer formation. In mutants, dauer formation is unaffected by growth factors such as decreased insulin/IGF-1 receptor function.
The inactivation of autophagy genes affects morphogenesis and survival. Certain autophagy genes have been shown to regulate cell development, with the effects mediated by insulin/IGF-1 or TGF signaling. Nematodes, for example, are unable to develop in large body size phenotype when autophagy gene activity is interrupted, as well as when insulin/IGF1 or TGF receptor activation is abnormal.
The p53 suppressor gene, which initiates autophagy, is mutated in around 50% of malignant malignancies. It has been revealed that P53 works as a crucial mediator for damage-induced apoptosis and has been found to promote autophagy in a DRAM- (Damage-regulated autophagy modulator-) dependent manner in human cancer cell lines to execute a complete cell death.
Autophagy is induced by nutrient restriction or rapamycin therapy. TORC1 (the rapamycin-sensitive TOR kinase complex 1) hyperphosphorylates Atg1, which has a reduced affinity for Atg1. Aside from that, TORC1 regulates the activation of various effectors that use phosphorylation to regulate the transcription or translation of certain proteins, some of which are essential for autophagy. TOR regulates induction of autophagy in cooperation with two other nutrient sensing pathways, that is, protein kinase A.
mTOR, coupled with insulin/IGF1 signaling, regulates autophagy at several levels. Normal cell development necessitates a careful balance of protein/organelle production and breakdown (turnover). Increased growth rate indicates the buildup of erroneous cytosolic components.
ROS (reactive oxygen species) are also involved in the regulation of starvation-induced autophagy. Atg4, an important protease in the autophagy process, has recently been identified as a direct target for oxidation by ROS. The accumulation of ROS produced during numerous biological functions, primarily respiration, results in oxidative stress. Cells respond to oxidative stress by activating numerous defensive mechanisms.
Several recent reviews of autophagy in relation to cancer and other disorders have been published. Autophagy is an evolutionarily conserved genetically designed mechanism that destroys long-lived cellular proteins and organelles. Autophagy is essential for normal development and responds to changing external stimuli.