This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
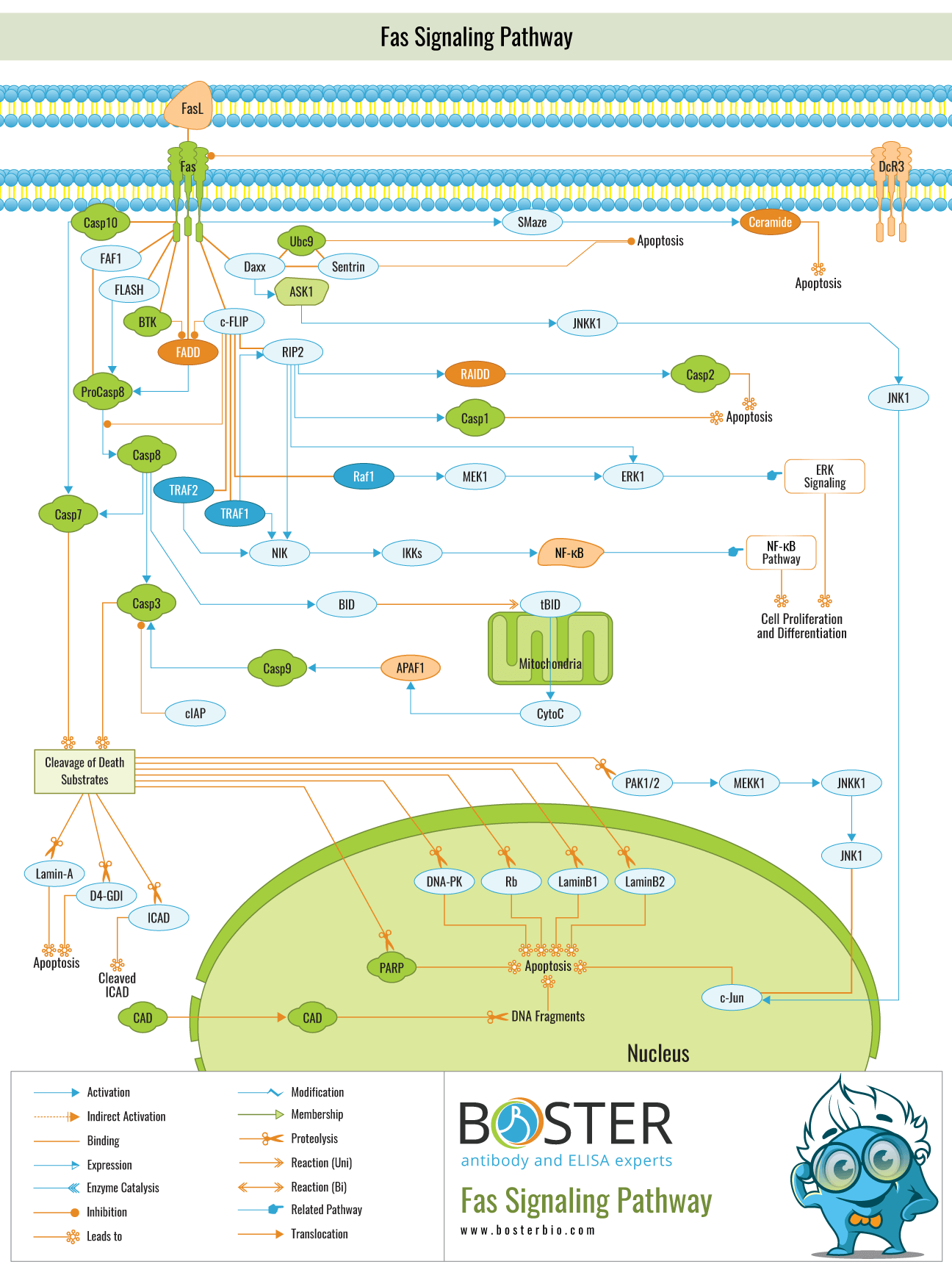
The Fas cell signaling pathway is critical for physiological regulation of programmed cell death (also known as apoptosis) and has been implicated in the pathogenesis of a variety of cancers and immune system diseases. Fas (also known as Apo1 or CD95) is a member of the TNFR (Tumor Necrosis Factor Receptor) superfamily that contains a death domain. Although the FasL (Fas Ligand)-Fas system has been lauded for its death-inducing function, it also transduces proliferative and activating signals via poorly defined pathways. By binding to FasL expressed on the surface of other cells, the Fas receptor induces an apoptotic signal.
Fas is a Type-I transmembrane protein, whereas FasL is a Type-II transmembrane protein belonging to the TNF family that can be shed in a soluble form via metalloproteinase action.
After binding to the FasL, the Fas receptor trimerizes and induces apoptosis via a cytoplasmic domain called the DD (Death Domain), which interacts with signaling adaptors such as FAF-1 (Fas-Associated Factor-1), FADD (Fas-Associated Death Domain), Daxx, FAP-1, FLASH (FLICE-associated huge), and RIP (Receptor-Interacting Protein). FADD contains a DED (Death Effector Domain) and recruits the inactive Procaspase-8 protein via homologous interaction. Additionally, this protein complex is referred to as DISC (Death-Inducing Signaling Pathways) and is found in Type-I cells. Procaspase-8 is activated proteolytically to form Caspase-8. Additionally, FADD aids in the activation of Caspase-10. Caspase-8 and Caspase-10, when activated, cleave and activate downstream effector caspases such as Caspase-3, -6, and -7. Caspase-8 activates Caspase-3 via two distinct mechanisms; the first is that Caspase-8 cleaves BID (Bcl2 Interacting Protein), and its COOH-terminal part translocates to mitochondria, where it induces the release of mitochondrial pro-apoptotic factors such as CytoC (Cytochrome-C) and SMAC (Second Mitochondria-derived Activator of Caspases), also known as CytoC is released and activates Caspase-9 when it binds to APAF1 (Apoplectic Protease Activating Factor-1) along with dATP and Procaspase-9. Caspase-9 catalyzes the cleavage of Procaspase-3 and the activation of Caspase-3. Another possibility is that Caspase-8 directly cleaves and activates Procaspase-3. Both pathways are regulated by the endogenous inhibitor FLIP (FLICE (FADD Like IL-1Beta-Converting Enzyme)-Inhibitory Protein), which may also be recruited by FADD. FLIP may also participate in a different signaling pathway, recruiting TRAF1 (Tumor Necrosis Factor-Associated Factor-1), TRAF2 (Tumor Necrosis Factor-Associated Factor-1), the MAPKKK (MAP Kinase Kinase Kinase Kinase Kinase) Raf1, and RIP to activate the ERK (Extracellular Signal-Regulated Kinase) and NF-KappaB This differential FLIP activity, which appears to reflect the activity of short versus long FLIP isoforms that promote death versus proliferation pathways, mediates a critical decision in response to Fas signaling: death versus proliferation/inflammation.
IAPs (Inhibitor of Apoptosis Proteins) inhibit caspases (2,3). Once activated, caspase-3, -6, and -7 degrade a variety of cytoskeletal and nuclear proteins (structural, signaling, and kinases) including GDID4 (GDP-Dissociation Inhibitor-D4), PARP (Poly ADP-Ribose Polymerase), Alpha-Fodrin, GAS2 (Growth Arrest Specific-2), Lamin-A and B (B1 and B2), and PAK PAK activity induced by caspase cleavage is required for JNK (c-Jun Terminal Kinase) activation induced by Fas receptor signaling, and thus PAK activity can also contribute to cell death induction. Caspase-3 also cleaves ICAD, a CAD inhibitor, allowing CAD to enter the nucleus and cleave DNA. Caspase-3 activation also cleaves DNA-PKcs in the nucleus, releasing the cleaved fragments into the cytosolic compartment. Additionally, stimulation of Fas results in significant changes in Rb (Retinoblastoma protein)
Apart from the FADD/Caspase-8 signaling cascade, the Fas receptor activates a variety of other signaling pathways. Daxx and RIP have been shown to bind to the FasR, modulating its signaling. Daxx (Death-Domain Associated protein) interacts with the Fas death domain despite the fact that Daxx itself lacks a DD. Daxx and FADD bind to Fas in distinct ways and activate distinct signaling pathways. Daxx promotes apoptosis mediated by Fas by activating the JNK kinase cascade, which results in the phosphorylation and activation of transcription factors such as c-Jun. Daxx is sequestered in the cytoplasm by the JNK kinase kinase ASK1 (Apoptosis Signal Regulating Kinase 1). Daxx interacts with and activates ASK1 in response to Fas stimulation. Daxx is found in the nucleus in the absence of ASK1, where it localizes to PODs (PML (Promyelocytic Leukemia Protein) Oncogenic Domains). ASK1 and Daxx can cause caspase-independent cell death that is unrelated to ASK1's kinase activity. ASK1 is also capable of inducing caspase-dependent apoptosis, but only when its kinase activity is activated. Daxx also interacts with sentrin, a ubiquitin-like protein that has the ability to covalently modify cellular proteins and functions as a Fas-binding protein that protects cells from Fas-induced cell death. Additionally, Daxx interacts with Ubc9, a critical protein that functions as a key-conjugating enzyme. The co-localization of Fas, sentrin, and Ubc9-binding regions suggests that this region is critical for Daxx regulation. Additionally, FLASH promotes Caspase-8 activation during Fas-mediated apoptosis.
Thus, it appears as though DED-containing proteins regulate the apoptotic process. FAF1 is a Fas-associating molecule that promotes apoptosis mediated by Fas. FAF1's N-terminus interacts with Fas's DD despite the absence of the typical death domain. FAF1 is a component of Fas-DISC, which is formed when the DED-like region of FAF1 interacts with the DEDs of Caspase-8 and FADD.