This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Toll-like receptors (TLRs) identify different molecular patterns associated with pathogens and are required for innate immune responses. They serve as the initial line of defense against pathogens infiltrating the body and are involved in inflammation, immune cell control, survival, and proliferation.
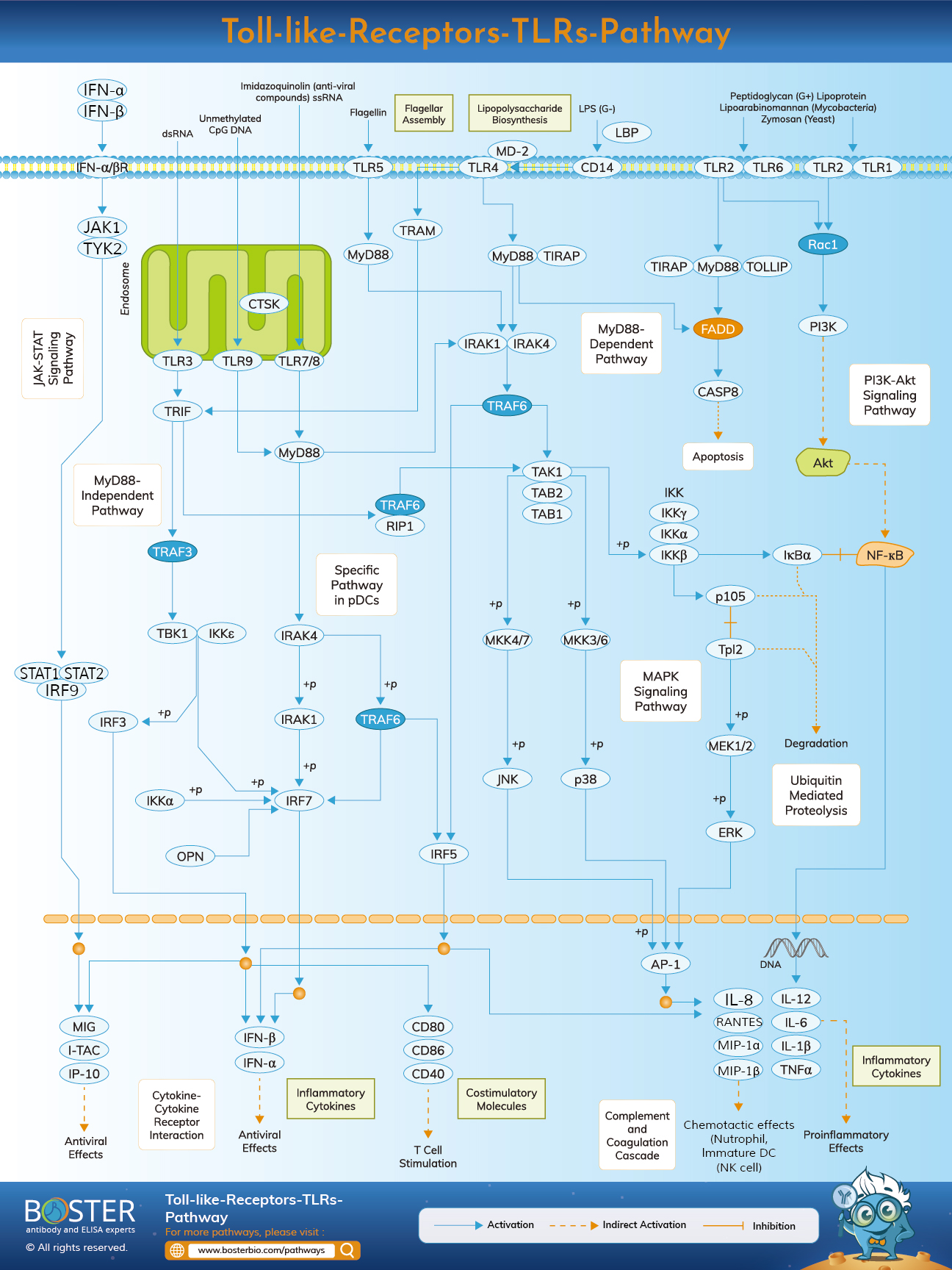
TLRs are a family of proteins that play a critical role in innate immunity. They are single domain transmembrane receptors that are often expressed in sentinel cells such as macrophages and dendritic cells, as well as a variety of other non-immune cells such as fibroblasts and epithelial cells. They recognize structurally conserved compounds generated from bacteria, dubbed pathogen-associated molecular patterns (PAMPs), or molecules originating from damaged cells, dubbed damage associated molecular patterns (DAMPs) (DAMPs). PAMPs include lipopolysaccharide (LPS), peptidoglycan (PGN), and lipopeptides from bacteria, as well as flagellin, bacterial DNA, and viral double-stranded RNA. DAMPs include both intracellular proteins such as heat shock proteins and extracellular matrix protein fragments. By inducing inflammatory cytokines, type I interferon (IFN), and other mediators, PRRs engage downstream signaling pathways that result in the development of innate immune responses.
Not only do these processes initiate acute host defensive responses, such as inflammation, but they also prime and regulate antigen-specific adaptive immune responses. These reactions are critical for eradicating invading microorganisms and instructing antigen-specific adaptive immune responses
The TLR family consists of ten members (TLR1–TLR10) in humans and twelve in mice (TLR1–TLR9, TLR11–TLR13). TLRs are found on the cell surface and in internal compartments such as the endosome, the endosome, and the lysosome. TLRs on the cell surface comprise TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10, whereas TLRs on the intracellular level include TLR3, TLR7, TLR8, TLR9, TLR11, TLR12, and TLR13. TLRs on the cell surface primarily recognize components of microbial membranes such as lipids, lipoproteins, and proteins. Intracellular TLRs identify nucleic acids generated from bacteria and viruses, as well as self-nucleic acids in autoimmune diseases.
The Toll-like receptor's function is typically based on the dimerization of two TLR molecules, but this is not always the case. For example, when TLR-1 and TLR-2 identify PAMPs molecules such as lipoproteins, peptidoglycans, lipotechoic acids (LTA, Gram-), zymosan, mannan, and tGPI-mucin, they form a dimer. When TLR-2 and TLR-6 recognize the same PAMPs listed above, they can also form a dimer. TLR-4 is capable of recognizing lipopolysaccharide (LPS, Gram+) and heterodimerizing with another TLR-4 molecule. TLR-5 are capable of recognizing bacterial flagellin but do not form a dimer. TLR-11 is functional in mice and is primarily involved in the recognition of uropathogenic microorganisms. TLR-3, 7, 8, 9, and 13 are expressed on the cytoplasmic surface of endosomes. TLR3 identifies double-stranded RNA (dsRNA) from viruses, small interfering RNAs (siRNAs), and self-RNA generated from injured cells. TLR-7 is mostly expressed on plasmacytoid dendritic cells (pDCs) and identifies viruses' single-stranded (ss) RNA. Additionally, it detects streptococcus B RNA in typical DCs (cDCs). TLR8 is activated in response to viral and bacterial RNA. TLR-9 identifies unmethylated CpG-DNA patterns in bacterial and viral DNA. TLR13 detects bacterial 23S rRNA and unidentified vesicular stomatitis virus components.
While there are numerous types of TLR molecules that identify a diverse array of ligands, all of these TLRs share a basic structural framework in their extracellular ligand-binding domains. Each of these domains is shaped like a horseshoe and is composed of leucine-rich repeat motifs. Upon ligand binding, two extracellular domains often form a "m"-shaped dimer, bringing the transmembrane and cytoplasmic domains into close proximity and initiating a downstream signaling cascade.
Sentinel cells such as macrophages use Toll-like receptors to detect pathogens via PAMPs such as LPS. LPS is a bacterial cell wall component. The process by which Toll-like receptors recognize lipopolysaccharides is complex and requires multiple accessory proteins. LPS-binding protein is a serum protein that binds to LPS monomers and transfers them to a protein named CD14. CD14 can be soluble or can form a glycosylphosphatidylinositol anchor on the cell surface. CD 14 transports and loads LPS into Toll-like receptors' extracellular domain. TLRs are capable of detecting LPS via an accessory protein called MD-2. Then, when LPS binds to the TLR-CD14-MD2 complex, homodimerization of TLRs occurs. Extracellular domain conformational changes initiate dimerization of the cytoplasmic Toll IL-1 receptor (TIR) domain. The conformational change in the TIR creates a novel scaffold for the binding of adaptor proteins to form a post-receptor signaling complex. The TIR contains a myeloid differentiation primary-response protein 88 (MyD88).
MyD88 acts as a bridge between TLRs/IL-1Rs and downstream signaling molecules that include DDs. It recognizes conformational changes in the TIR domain of TLRs, attaches to the new receptor complex, and transmits signals via contact with IL-1R-associated kinases via the amino (N)-terminal death domain (DD) (IRAKs). As a result, a complicated cascade of signaling events occurs, alerting the cell to the presence of a pathogen. There are a total of four IRAKs (IRAK 1, 2, 4, M). They have an N-terminal DD and a serine/threonine kinase domain in the center. IRAK1 and IRAK4 have intrinsic kinase activity, whereas IRAK2 and IRAK-M do not. MyD88 activated IRAK4, which continued to activate IRAK1. IRAK1 then activates TRAF6. TRAF6 is a member of the tumor necrosis factor receptor (TNFR)-associated factor (TRAF) family of proteins involved in cytokine signaling. TRAF6 is recruited to the receptor complex and activated by IRAK-1, which binds to TRAF6's TRAF domain. The IRAK-1/TRAF6 complex then dissociates from the receptor and binds with TGF-beta-activated kinase 1 (TAK1) and TAK1-binding proteins TAB1 and TAB2, respectively. TRAF6, TAK1, TAB1, and TAB2 create a big complex with additional proteins in the cytoplasm, including the E2 ligases Ubc13 and Uev1A.
The Ubc13 and Uev1A complex has been demonstrated to catalyze the formation of a TRAF6 Lys 63-linked polyubiquitin chain, thereby activating TAK1 and ultimately NF-kB via TRAF6. These signaling pathways outlined above are referred to as MyD88-dependent pathways due to the fact that the signal originates from the MyD88 protein. There is also a mechanism known as the MyD88-independent pathway, in which signaling does not begin with MyD88. Rather than that, the signal originates with the TRIF protein. TRAF6 and TRAF3 interact with TRIF. TRAF6 recruits the kinase RIP-1, which interacts with and activates the TAK1 complex, resulting in the activation of NF-kB and MAPKs, as well as the production of pro-inflammatory cytokines. TRAF3, on the other hand, recruits the IKK-related kinases TBK1 and IKKi, as well as NEMO, to phosphorylate and activate IRF3. IRF3 dimersizes and translocates from the cytoplasm to the nucleus, where it stimulates the production of type I IFN.
Naturally, there is some negative control by a variety of molecules via a variety of pathways to avoid or terminate excessive immune responses associated with autoimmunity and inflammatory illness. ST2825, SOCS1, and Cbl-b inhibit the activation of the MyD88-dependent pathway, whereas SARM and TAG inhibit the activation of the TRIF-dependent route. These compounds form complexes with MyD88 or TRIF in order to inhibit their binding to TLRs or downstream molecules. SOCS3 and DUBA both inhibit TRAF3 activation.
TRAF6 is inhibited by a variety of molecules, including A20, USP4, CYLD, TANK, TRIM38, and SHP. TRIM30a and A20 suppress TAK1 activation.