This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
In epigenetic research, protein–DNA interactions (such as histone modifications and transcription factor binding sites) are central to understanding the regulation of gene expression. Chemical modifications of histones can alter chromatin conformation, placing chromatin in either an open or closed state, thereby regulating the accessibility of transcription factors to DNA. For example, specific histone modifications can promote chromatin opening, allowing regulatory factors such as transcription factors to access DNA. Accordingly, techniques for studying protein–DNA interactions can reveal these regulatory mechanisms.
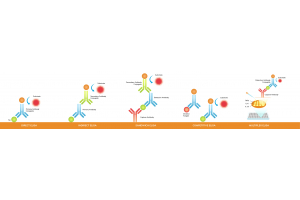
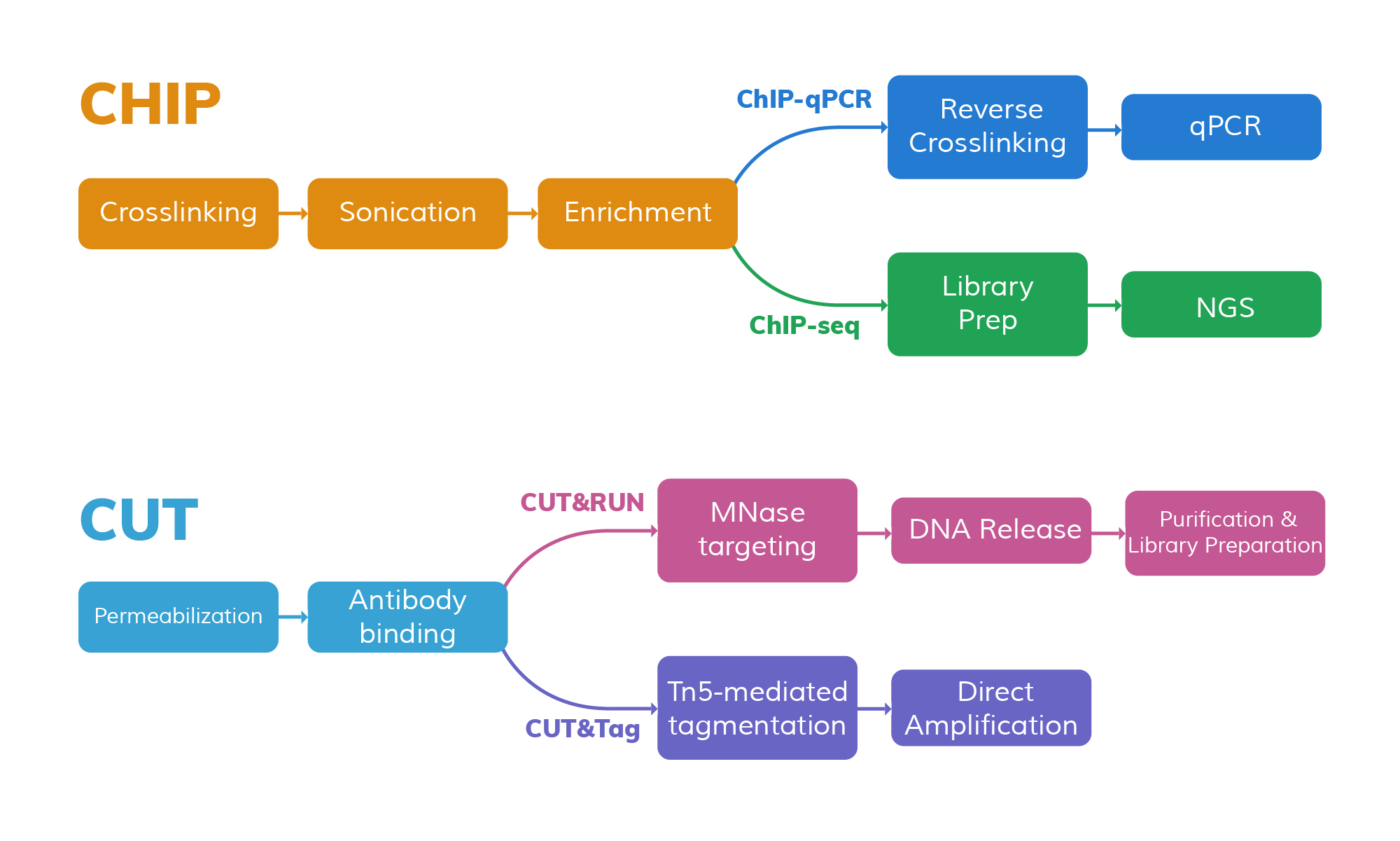
Chromatin immunoprecipitation (ChIP) is a classic method in this field. It uses specific antibodies to enrich for target protein–DNA complexes, thereby capturing the binding sites between particular proteins and DNA. With the development of high-throughput sequencing technologies, ChIP-based methods such as ChIP-seq have become standard tools for generating genome-wide maps of protein–DNA interactions.
In recent years, new techniques such as CUT&RUN (Cleavage Under Targets and Release Using Nuclease) and CUT&Tag (Cleavage Under Targets and Tagmentation using Tn5 transposase) have been introduced to improve sensitivity, reduce required sample input, and significantly lower background noise. These techniques have now become core tools in epigenetic research, providing strong technical support for both scientific discovery and clinical translation.
ChIP-qPCR is based on the principle of chromatin immunoprecipitation. First, protein–DNA complexes are usually crosslinked in vivo with formaldehyde. Then, cells are lysed and chromatin is sheared by sonication to fragment DNA (typically 200–600 bp).
Next, a primary antibody specific to the target protein (such as a transcription factor or a modified histone) is used to bind the protein–DNA complexes. The antibody complexes are then enriched using Protein A/G magnetic beads. Afterward, proteins are eluted, and crosslinks are reversed to recover the DNA.
Finally, qPCR is performed on regions of interest in the genome (such as known promoters or enhancers) to quantify the binding abundance of the target protein in that region.
ChIP-qPCR has advantages including high sensitivity, good signal-to-noise ratio, and the ability to quantitatively compare binding intensities at specific sites under different conditions.
However, its limitations include only being able to detect known sites (not genome-wide), the need for specific primer design, and a relatively high input requirement (typically 10⁴ to 10⁶ cells) to obtain a detectable signal.
ChIP-seq builds upon the idea of ChIP-qPCR and uses the same crosslinking and immunoprecipitation steps to obtain DNA fragments bound by the target protein. Then, high-throughput sequencing is used to sequence all enriched DNA fragments.
The workflow includes: crosslinking, cell lysis, chromatin shearing, antibody-based immunoprecipitation, reversing crosslinks and purifying DNA, adaptor ligation, PCR amplification of the sequencing library, and finally sequencing.
Unlike ChIP-qPCR, ChIP-seq can simultaneously determine the positions of all enriched fragments across the genome, revealing the global distribution of protein–DNA binding.
ChIP-seq features high resolution (tens to hundreds of base pairs) and genome-wide coverage. However, it requires a large number of cells (typically millions) to generate high-quality data.
Additionally, ChIP-seq involves many steps (crosslinking, shearing, IP, library construction, etc.), making it complex and time-consuming (usually 4–7 days).
Due to non-specific binding and sample loss, ChIP-seq also has relatively high background noise and requires careful optimization and control samples.
In summary, ChIP-seq is a classical tool for studying protein–DNA interactions and is suitable for mapping genome-wide binding sites of transcription factors and histone modifications.
CUT&RUN is a native chromatin profiling technique that does not require crosslinking. It uses a target-specific antibody and micrococcal nuclease fused to Protein A/G (pA-MNase) to cleave DNA near protein-binding sites inside the nucleus, thereby efficiently releasing the bound fragments.
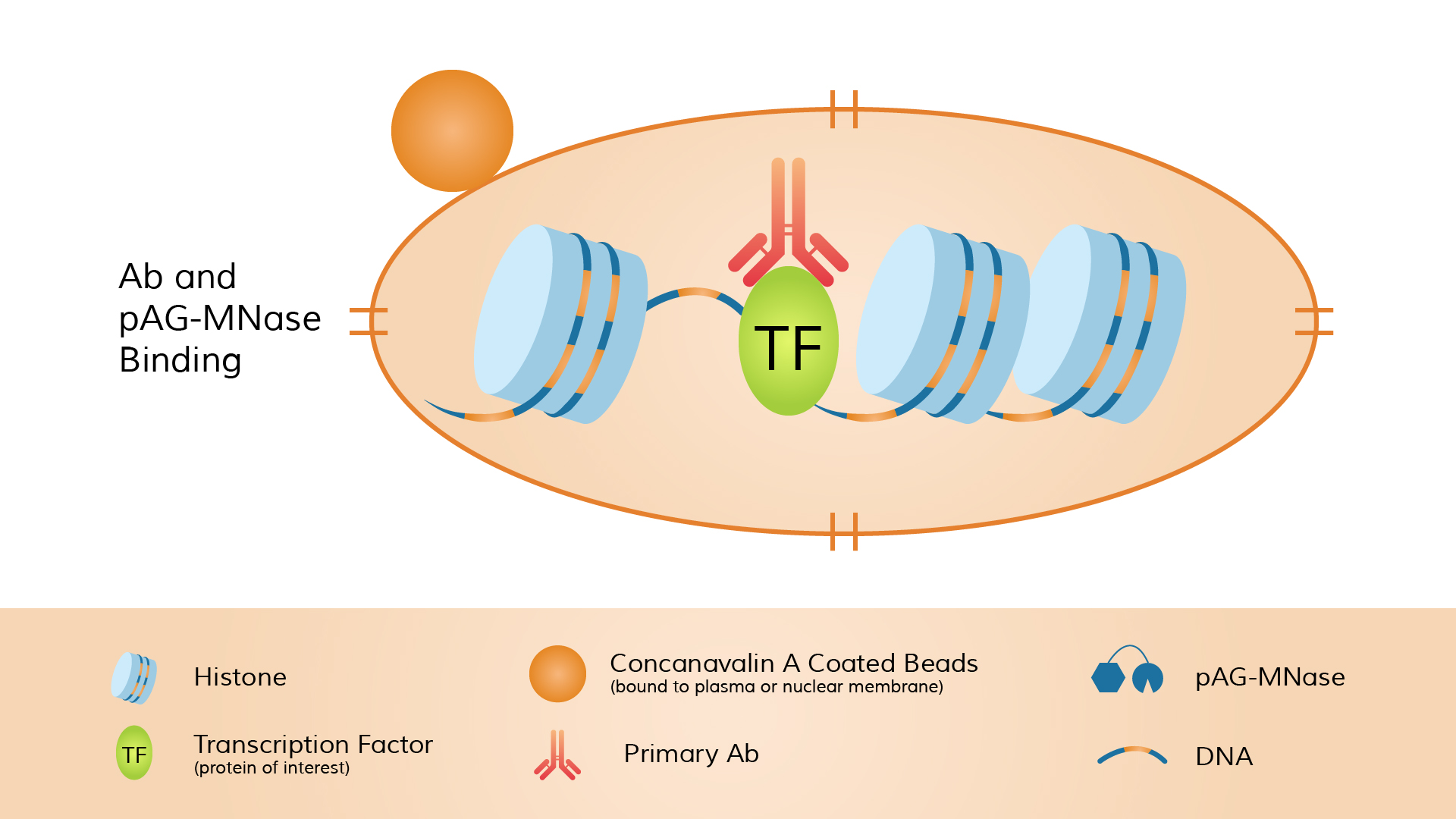
The typical workflow includes: immobilizing unfixed cells or nuclei onto magnetic beads, incubating with a primary antibody targeting the nuclear protein of interest (which specifically binds the target), and then adding pA-MNase.
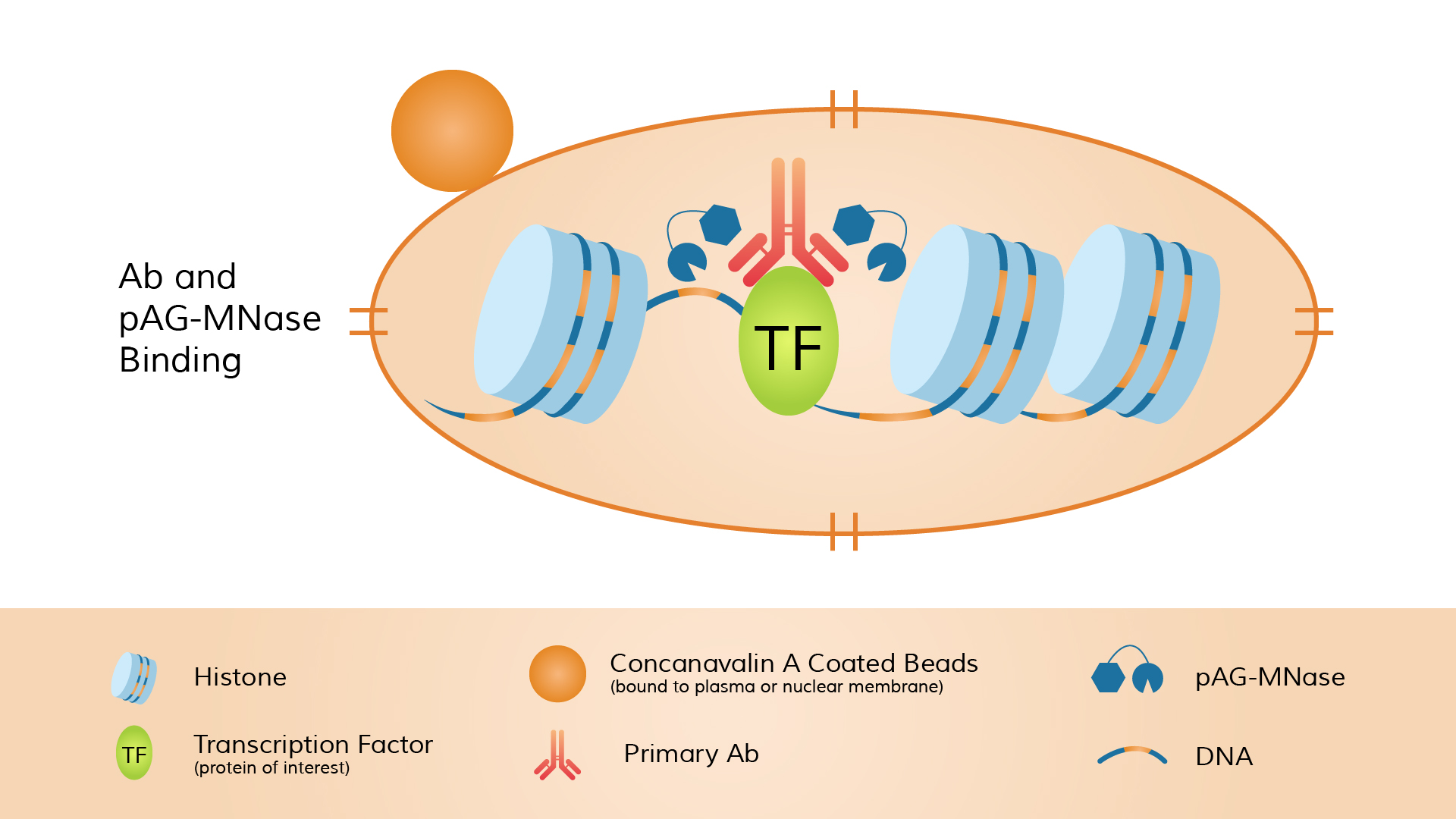
The enzyme binds to the antibody (via Protein A binding to the Fc region of the antibody), forming an antibody–pA-MNase–protein complex.
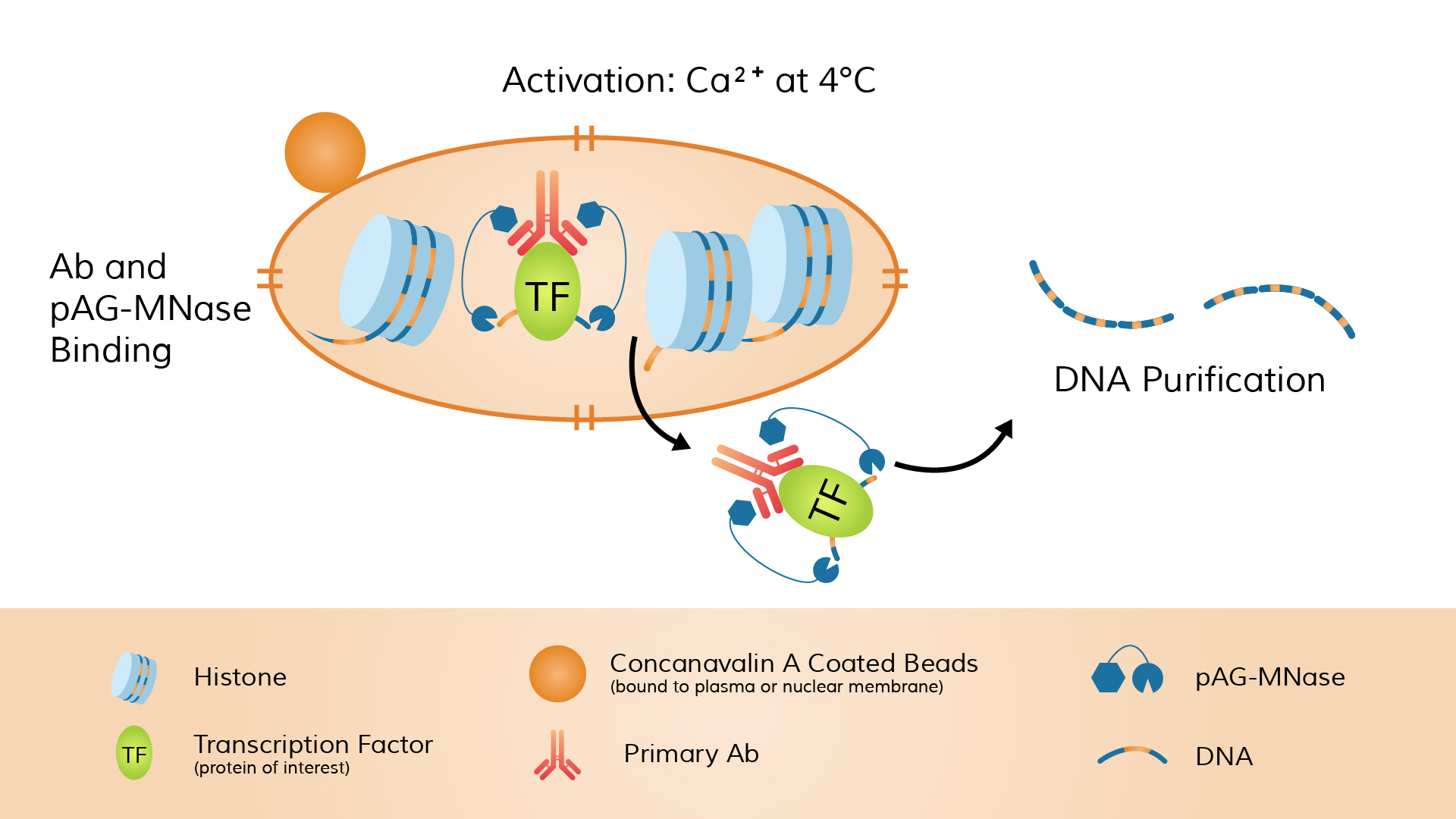
Calcium ions (Ca²⁺) are then added to activate MNase cleavage, allowing the enzyme to cut DNA near the protein-binding sites and release the protein–DNA fragments from chromatin.
After cleavage, the DNA fragments are separated and purified from the solution and then subjected to high-throughput sequencing.
Unlike traditional ChIP, CUT&RUN does not require sonication or immunoprecipitation, and the entire reaction is completed under native chromatin conditions.
As a result, CUT&RUN requires fewer starting cells (as few as 10³–10⁵), offers high specificity, and produces very low background noise.
A standard workflow can be completed within 1–2 days.
It’s important to note that in CUT&RUN, the antibody and pA-MNase are added separately (i.e., the enzyme is not pre-bound to the antibody) to ensure that cleavage occurs only near the antibody-marked targets.
In summary, CUT&RUN enables the direct enzymatic release of DNA fragments bound to the target protein, achieving higher sensitivity and a better signal-to-noise ratio.
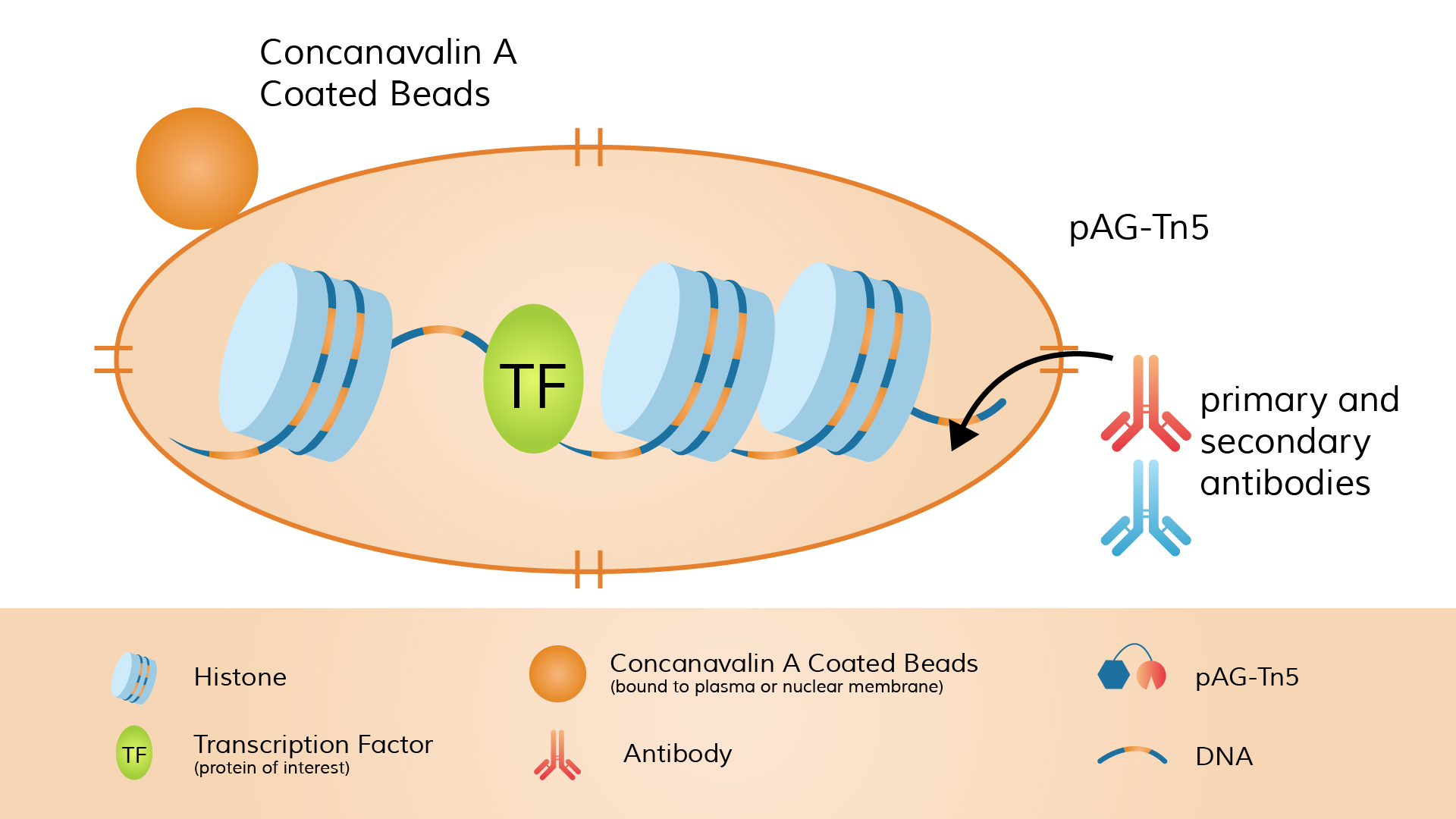
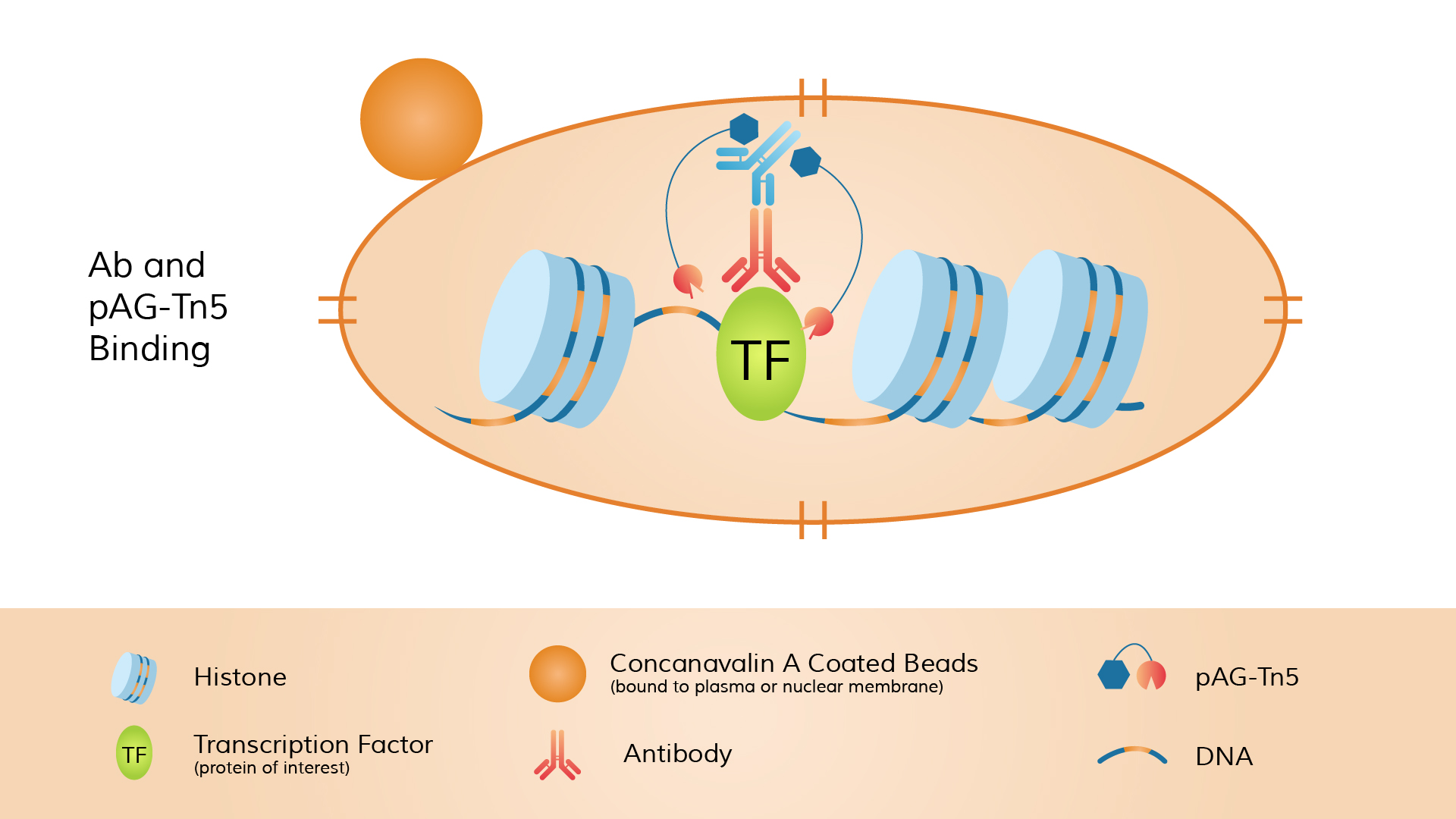
CUT&Tag (Cleavage Under Targets & Tagmentation) is also an in situ chromatin profiling method. Its distinguishing feature is that the Tn5 transposase is “tethered” to the antibody complex, enabling simultaneous DNA cleavage and sequencing adapter insertion at the target site. The procedure begins with a primary antibody targeting the protein of interest or histone modification. A secondary antibody (e.g., anti-rabbit or anti-mouse) is then added to amplify the signal.
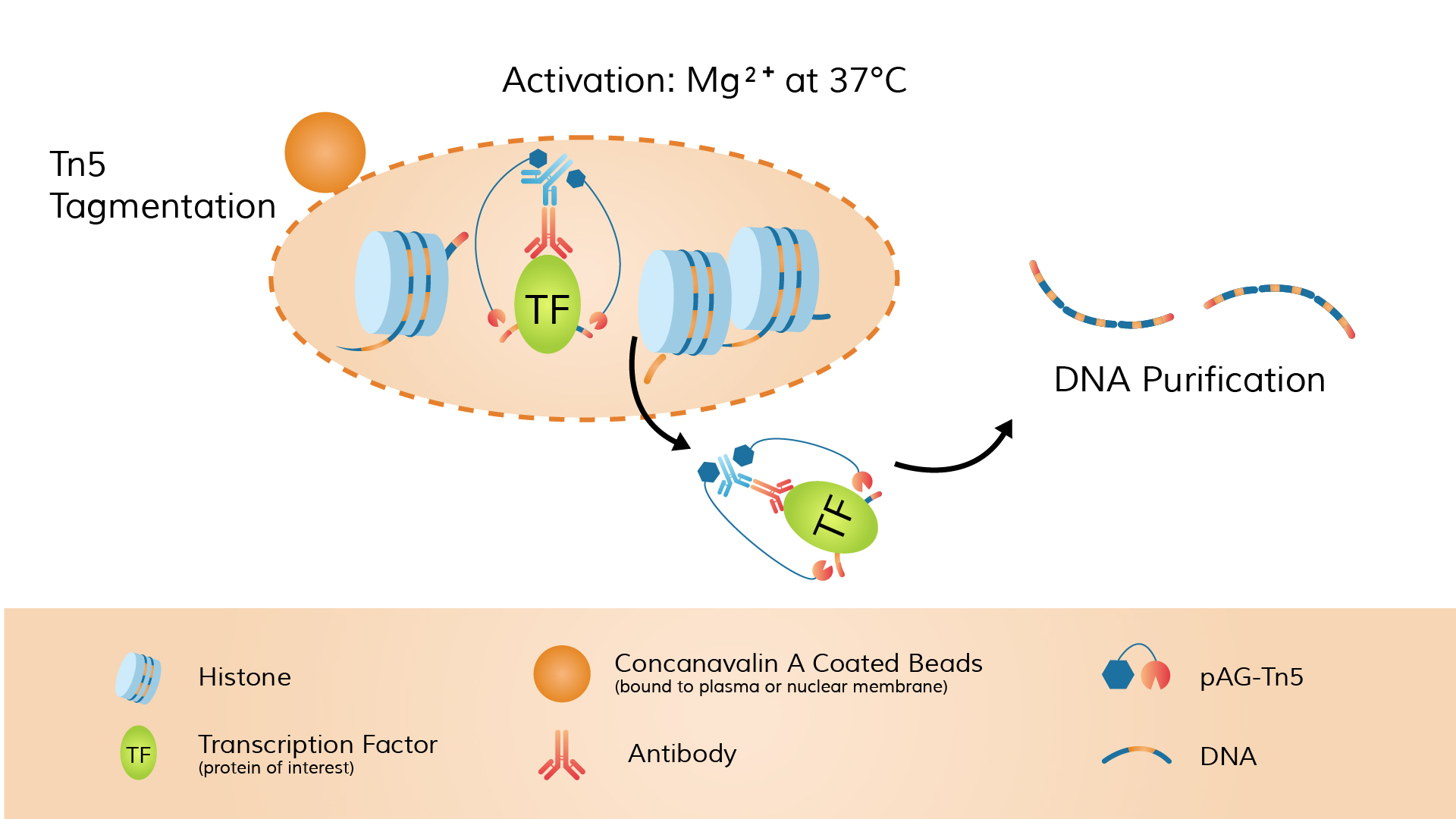
Next, a fusion protein of pA-Tn5 transposase preloaded with double-stranded sequencing adapters (called the pA-Tn5 adapter-transposome) is introduced. The Protein A domain of pA-Tn5 binds to the Fc region of the antibody. Once pA-Tn5 is localized to the antibody-labeled site, it simultaneously cleaves DNA and inserts sequencing adapters in the presence of Mg²⁺.
Unlike CUT&RUN, which uses MNase, CUT&Tag employs the Tn5 transposase. Furthermore, the Tn5 used in CUT&Tag is pre-loaded with sequencing adapters before the reaction. Therefore, CUT&Tag can fragment DNA and integrate adapters in one step, without additional end-repair or adapter ligation steps.
The advantages of CUT&Tag include requiring extremely low input—usable data can be obtained from as few as 10⁴ or fewer cells. The workflow is relatively streamlined and can be completed within 1–2 days, while producing data with an extremely high signal-to-noise ratio. CUT&Tag is commonly used to detect chromatin marks such as histone modifications.
It should be noted that some studies suggest CUT&Tag may be less effective than CUT&RUN for profiling chromatin-binding proteins such as certain transcription factors. This difference may be related to enzyme activity or target accessibility. Overall, CUT&Tag offers significant advantages in experiments with minimal sample availability, tight timelines, or high-throughput requirements.
| Category | ChIP-qPCR | ChIP-seq | CUT&RUN | CUT&Tag |
|---|---|---|---|---|
| Starting Material | High (typically 10⁴–10⁶ cells) | Very high (millions of cells) | Low (10³–10⁵ cells) | Extremely low (10³–10⁴ cells; single-cell possible) |
| Peak Resolution | Medium (depends on chromatin fragmentation, usually several hundred bp) | High (tens to over a hundred bp) | Very high (precise MNase cleavage, down to single-digit bp) | Very high (precise Tn5 insertion, down to single-digit bp) |
| Operational Complexity (Time) | High (requires crosslinking, shearing, IP; takes several days) | Very high (~1 week; includes multiple steps) | Moderate (no crosslinking; ~1–2 days) | Low (simplified in situ protocol; ~2 days) |
| Background Noise | Relatively high (requires antibody and elution optimization) | Relatively high (many non-specific bindings) | Very low (in situ cleavage minimizes background) | Extremely low (adaptors inserted directly at target sites) |
| Library Construction Method | No full library; qPCR of specific loci | Traditional library prep (end repair, adaptor ligation, PCR) | End repair + adaptor ligation + PCR | One-tube PCR using Tn5-inserted adaptors |
As shown in the table, although ChIP-qPCR and ChIP-seq have been widely used, they require a large amount of input material (ChIP-seq typically requires ≥10⁶ cells), and the procedures are complex with relatively high background noise. In contrast, both CUT&RUN and CUT&Tag are performed in situ, eliminating or reducing the need for crosslinking and complicated elution steps, which significantly shortens the experimental time and reduces background signals.
Among them, CUT&Tag further simplifies library preparation (by preloading sequencing adapters on Tn5), providing clear advantages in terms of input requirements and signal-to-noise ratio. However, there are slight differences in the types of target proteins between the two: CUT&Tag is well-suited for profiling histone modifications and similar chromatin marks, but may be less stable than CUT&RUN when targeting certain transcription factors.
ChIP-seq’s main advantage lies in its maturity and reliability, with compatibility across a wide range of targets and sample types, though it comes with higher costs and the need for high-throughput sequencing. Overall, these four techniques differ fundamentally in sample requirements, technical complexity, and data quality. Researchers should carefully choose among them based on the specific aims of their experiment.
Best suited for validating and quantifying protein–DNA interactions at known genomic loci. It is particularly effective for comparing binding strength at specific sites under different experimental conditions. However, due to the requirement for specific primer design and the limitation to a small number of targets, ChIP-qPCR is not appropriate for genome-wide exploration. It is mainly used for candidate site verification or small-scale studies.
Ideal for genome-wide profiling of
protein–DNA binding sites, such as identifying unknown transcription factor
binding motifs or histone modification regions. ChIP-seq requires a large
number of cells and is commonly used in studies demanding high-throughput
coverage, including analyses of epigenomic changes during cell differentiation
or in disease models. Due to its relatively low signal-to-noise ratio, it
demands robust control experiments and extensive bioinformatics post-processing
to filter background noise.
Highly suitable for rare samples or experiments with limited cell input (as few as 10³–10⁵ cells can yield high-quality data). CUT&RUN is effective for high-resolution mapping of transcription factors—especially chromatin-associated proteins—and histone modifications. The in situ cleavage mechanism results in very low background noise, making it ideal for applications that require high signal clarity. Its simplicity and short processing time also make it particularly useful for quick validations or repeated assays.
Due to the activity of Tn5 transposase, CUT&Tag can generate high-quality data from ultra-low cell inputs (10³–10⁴ cells, or even single cells). It excels at profiling histone modifications such as methylation and acetylation, as these targets typically have high antibody efficiency and are readily cleaved. However, for loosely bound transcription factors, CUT&RUN is often recommended instead. The overall workflow of CUT&Tag is faster (approximately 1–2 days), making it well-suited for large-scale sample screening, time-sensitive experiments, and automation platforms. CUT&Tag is also compatible with single-cell epigenomic sequencing (scCUT&Tag), enabling analysis of cellular heterogeneity and rare cell populations.
For large-scale transcription factor screening or chromatin conformation studies, ChIP-seq or CUT&RUN may be considered depending on the experimental context. For example, to investigate 3D genome structures such as enhancer–promoter loops, derived methods like HiChIP may be used (see next section). Additionally, if the goal is simply to identify open chromatin regions rather than protein binding, ATAC-seq and its single-cell version would be more appropriate.
Proposed by Japanese researchers in 2018, ChIL-seq replaces traditional immunoprecipitation (IP) with immunofluorescent labeling, thereby significantly reducing sample loss. In this method, unfixed cells are first incubated with a primary antibody targeting the protein of interest, followed by a secondary antibody that chemically crosslinks and recruits Tn5 transposase near the target site. This enables simultaneous cleavage and adapter insertion at the target locus. The method eliminates the need for immunoprecipitation, drastically lowering the input requirement: studies have shown that ChIL-seq can detect histone modifications and DNA-binding factors with as few as 100–1,000 cells—and even down to the single-cell level. This makes ChIL-seq highly suitable for rare cell types or extremely limited samples. It also supports in situ amplification before lysis, which is advantageous for adherent cells. However, ChIL-seq has some limitations, such as lower sensitivity in heterochromatin regions and a relatively long experimental duration (3–4 days). Overall, ChIL-seq provides a feasible approach for capturing protein–DNA interactions from very low input material and is particularly well-suited for single-cell epigenomic studies.
HiChIP integrates the concepts of in situ Hi-C and ChIP to map long-range chromatin interactions mediated by specific proteins. The protocol begins with an in-nucleus Hi-C process—crosslinking, restriction enzyme digestion, end repair and biotin labeling, and proximity ligation—followed by cell lysis and immunoprecipitation to enrich DNA fragments associated with the protein of interest. Sequencing is then performed on biotin-tagged fragments, with each read pair representing two distant genomic regions brought into proximity via the protein of interest. Compared to traditional ChIA-PET, HiChIP significantly improves data yield (with over 10-fold more usable reads) and dramatically reduces the required cell input. Since HiChIP proceeds after in situ crosslinking, it also decreases false positives and improves signal-to-noise ratio. Typically completed within two days, HiChIP is well-suited for studying chromatin architecture mediated by transcription factors or structural proteins like CTCF or Cohesin. It has been applied to explore physical interactions between regulatory elements, transcriptional mechanisms, and how histone modifications influence gene expression.
While scATAC-seq primarily identifies open chromatin regions rather than direct protein–DNA interactions, it has become a key technique in epigenetic profiling. Using microfluidics or barcoded transposase strategies, scATAC-seq marks accessible chromatin at the single-cell level through Tn5 transposition. Its major strength lies in resolving chromatin accessibility within complex tissues or heterogeneous cell populations, enabling the identification of regulatory elements specific to individual subpopulations. For instance, scATAC-seq can reveal cellular heterogeneity in tumors or during developmental processes. However, it requires a relatively high number of input cells, costly barcoding reagents, and complex downstream data analysis—making it significantly more demanding and expensive than bulk ATAC-seq. In summary, scATAC-seq is a powerful tool for studying cell-to-cell variability and fate decisions but is primarily focused on chromatin accessibility rather than direct protein–DNA interactions.
Each of these emerging techniques offers unique strengths and limitations. For example, HiChIP is ideal for studying 3D genome structures mediated by specific proteins; ChIL-seq and CUT&Tag are suitable for ultra-low input or single-cell applications; and scATAC-seq is best for profiling genome-wide chromatin accessibility. As technological innovation continues, the toolkit for epigenetic research is becoming increasingly diverse and refined—offering researchers richer perspectives and more robust data to unravel the complexities of gene regulation.