This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
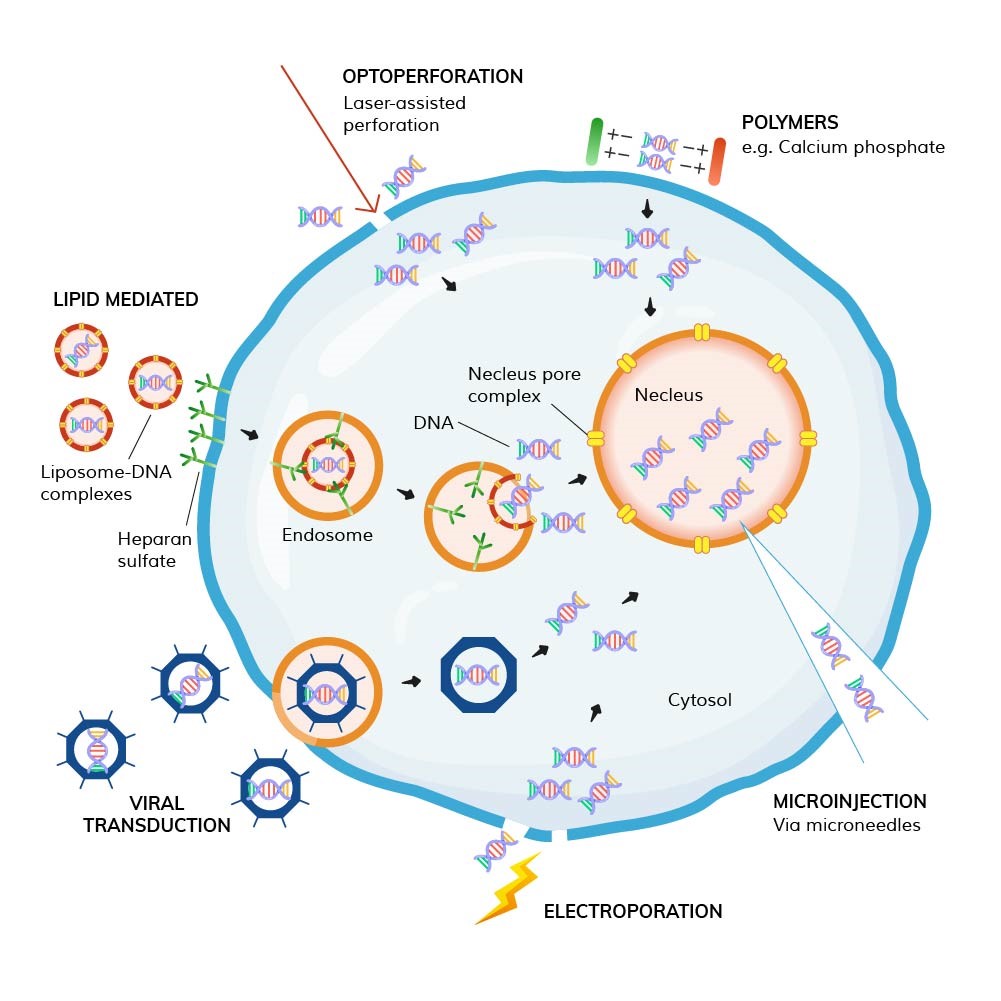
Transfection is the process of introducing exogenous genetic material, such as DNA or RNA, into eukaryotic cells. This exogenous genetic material can be artificially synthesized gene fragments or gene sequences extracted from other organisms. Through transfection, researchers can artificially alter the genetic information inside the cells, enabling the study of gene functions, protein production, gene therapy development, cell signaling pathways, and disease mechanisms, among others.
Principle:
Cells have an inherent barrier function, where the cell membrane prevents external substances from entering the cell, thereby maintaining the stability of the internal environment. The core of transfection technology is to break through this barrier of the cell membrane and successfully deliver exogenous genetic material into the cell.
Classification:
1. Chemical Transfection
Liposome-mediated Transfection: One of the most commonly used chemical transfection methods. Liposomes are tiny vesicles composed of a phospholipid bilayer that can fuse with the cell membrane. The exogenous DNA or RNA is encapsulated inside the liposome, forming a liposome-nucleic acid complex. Cells internalize this complex via endocytosis, and the nucleic acid is released from the liposome into the cytoplasm. Liposome-mediated transfection is relatively simple, has a high transfection efficiency, and is suitable for a variety of cell types. However, it may exhibit some toxicity to certain cells, and transfection efficiency is influenced by factors such as the liposome-nucleic acid ratio and cell density.
Cationic Polymer Transfection: Cationic polymers carry a positive charge, which allows them to bind with negatively charged nucleic acids through electrostatic interactions, forming stable complexes. These complexes can adsorb to the cell surface and enter cells via endocytosis. Compared to liposome transfection, cationic polymer transfection has better cell adaptability and can achieve good results with some difficult-to-transfect cell types, but it may also pose cell toxicity issues.
Calcium Phosphate Precipitation: This method involves mixing exogenous DNA with calcium phosphate, utilizing the cell membrane's ability to adsorb exogenous DNA and the cytoplasm's ability to uptake DNA, enabling the entry of DNA into the cell.
2. Physical Transfection
Electroporation: After mixing the cell suspension with exogenous nucleic acids, the mixture is placed in an electroporation device, where a brief high-voltage pulse is applied. The electric pulse temporarily forms reversible pores in the cell membrane, through which exogenous nucleic acids can enter the cell. Electroporation offers high transfection efficiency, particularly for difficult-to-transfect cells. However, the process can damage cells, affecting their survival and growth.
Microinjection: This technique uses microscopic manipulation to directly insert fine needle into the cell and inject exogenous nucleic acids into the cell. This method allows for precise delivery of genetic material into specific cells, with high transfection efficiency and minimal impact on cells. However, it is technically challenging and has low throughput, making it unsuitable for large-scale transfection experiments.
Gene Gun: This technique uses high-speed particles to deliver exogenous DNA or RNA into target cells, thereby achieving gene introduction.
3. Biological Transfection
Virus-Mediated Transfection: Common viral vectors include retroviruses, lentiviruses, and adenoviruses. These viruses can efficiently infect cells and integrate their genetic material into the host cell's genome (retroviruses and lentiviruses) or express the material transiently in the host cell (adenoviruses). Virus-mediated transfection has the advantage of high transfection efficiency and the ability to infect a wide variety of cell types, making it especially suitable for in vivo gene delivery. However, the preparation of viral vectors is complex and presents potential biosafety risks, such as immune responses caused by viral infection.
Transfection Methods: Transient vs. Stable Transfection
| Project | Transient Transfection | Stable Transfection |
|---|---|---|
| Genetic inheritance | Exogenous genes are not passed to offspring | Exogenous genes are passed to offspring |
| Integration site | Free in the cytoplasm or nucleus | Integrated into the cell genome |
| Screening steps | No need for screening | Requires selective pressure (e.g., antibiotics) |
| Experiment cycle | Quick, usually completed in a few days | Slow, may take weeks to months |
| Cost | Low experiment cost | High experiment cost, but costs lower after producing stable clones |
| Purpose | Quick gene expression analysis, protein expression, functional analysis | Long-term studies, drug screening, cell line establishment |
| Common methods | Liposome transfection, calcium phosphate precipitation, electroporation, etc. | Lentivirus and retrovirus transfection, etc. |
Prepare the foreign DNA (e.g., plasmid) to be transfected; prepare an appropriate lipofection reagent; prepare serum-free medium, complete medium containing serum and antibiotics; prepare reagents for cell treatment such as trypsin and PBS buffer.
Prepare the sterile workbench, CO₂ incubator, centrifuge, microscope, pipettes with tips, cell culture plates, and other experimental instruments and consumables, ensuring that all equipment is functioning correctly and the consumables are sterilized.
One day before transfection, seed 293T cells at a density of 3×10⁵-5×10⁵ cells per well in a 6-well plate, ensuring that the cells reach 50%-80% confluence at the time of transfection. Incubate the cells at 37°C in a 5% CO₂ incubator.
In a sterile environment, prepare 2 µg of plasmid DNA per well and add it to 100 µL of serum-free medium. Gently mix using a vortex mixer or pipette and pipette 15-20 times to ensure the plasmid DNA is fully dissolved and evenly dispersed.
Add 2 µL of transfection reagent directly into the DNA dilution, gently mix again with the vortex mixer or pipette 15-20 times. Let the mixture stand at room temperature for 10-20 minutes to allow DNA to fully bind to the liposomes and form a complex.
| Culture Container | DNA Amount (per well) | Transfection Reagent Amount (per well) | Dilution Volume | Medium Volume |
|---|---|---|---|---|
| 96-well plate | 0.1 µg | 0.1 µL | 10 µL | 100 µL |
| 48-well plate | 0.2 µg | 0.2 µL | 20 µL | 200 µL |
| 24-well plate | 0.5 µg | 0.5 µL | 50 µL | 500 µL |
| 12-well plate | 1 µg | 1 µL | 50 µL | 1 mL |
| 6-well plate | 2 µg | 2 µL | 100 µL | 2 mL |
Remove the culture plate from the incubator, discard the original medium, and gently wash the cells 1-2 times with PBS buffer to remove residual serum and metabolites. Then add an appropriate amount of serum-free medium to create a serum-free environment, which helps the lipid-DNA complex bind to the cells.
Slowly add the prepared DNA-lipid complex to the culture plate, gently shake the plate to ensure even distribution of the complex on the cell surface. Return the plate to the CO₂ incubator for continued incubation.
12-18 hours after transfection, discard the serum-free medium containing the complex, and add fresh complete medium containing serum and antibiotics. Continue to culture the cells. At this stage, the cells begin to take up and express the foreign DNA.
Continue to culture the cells for another 24 hours after medium replacement to allow sufficient expression of the target gene inside the cells.
Use a fluorescence microscope to observe the cells (if the plasmid carries a fluorescent marker like GFP) to assess transfection efficiency. The optimal time for observation is typically 24-48 hours post-transfection when the target gene expression is high and stable.
1. Fluorescence Marker Method:
If the foreign DNA carries a fluorescent marker (e.g., GFP), use a fluorescence microscope to directly observe the fluorescence expression in the cells. Randomly select multiple fields of view and calculate the ratio of fluorescent-positive cells to total cells to evaluate transfection efficiency. For example, if 800 out of 1000 cells exhibit green fluorescence in 10 fields, the transfection efficiency is 80%. Flow cytometry can also be used for large-scale analysis, providing a more accurate measure of transfection efficiency and enabling analysis of cell population heterogeneity.
2. Reporter Gene Detection:
When using plasmids containing reporter genes (e.g., luciferase gene), the transfection efficiency can be evaluated by detecting the activity of the reporter gene. As an extension, researchers may use our Reporter cell lines to measure promoter activation via luciferase for functional insight. After collecting the transfected cells, process them according to the luciferase assay kit’s instructions and measure the luciferase activity using a microplate reader. The higher the activity, the higher the transfection efficiency.
1. RT-PCR:
Extract total RNA from transfected cells, reverse transcribe it into cDNA, and then amplify the target gene using PCR.Analyze the PCR products using agarose gel electrophoresis and compare the band intensity with that of the internal control gene for semi-quantitative analysis of the target gene expression. If the target gene band is significantly brighter than the control group, it indicates successful expression of the foreign gene with a high expression level.
2. Western Blot:
Extract total protein from the cells and separate the proteins using SDS-PAGE electrophoresis. Transfer the proteins to a membrane and use specific antibodies to bind the target protein. Detect the target protein’s expression via chemiluminescence or colorimetric reaction. If a specific band appears in the transfection group and its intensity is higher than the control group, it indicates successful expression and an increased amount of the target protein.
1. Cell Proliferation Assay:
Use MTT or CCK-8 assays to detect cell proliferation after transfection. At different time points (e.g., 24, 48, 72 hours post-transfection), add the corresponding detection reagent to the culture plate, incubate for a certain period, and measure the absorbance value using a microplate reader. Plot a cell growth curve based on the absorbance values to analyze the impact of foreign gene transfection on cell proliferation. If the transfected cells show a steeper growth curve than the control group, it indicates that the foreign gene promotes cell proliferation.
2. Cell Apoptosis Assay:
Use flow cytometry combined with Annexin V-FITC/PI double staining to detect cell apoptosis. Collect the transfected cells, stain them, and detect apoptosis using a flow cytometer. If the apoptosis rate of the transfected cells differs significantly from that of the control group, it indicates that the transfection of the foreign gene affects cell apoptosis.
The success of transfection experiments not only depends on following standardized experimental protocols but also on accurate result analysis. Through analysis of transfection efficiency, gene expression, and cell phenotypes, the mechanism of action of the foreign gene in the cells can be deeply explored, providing valuable data support for life sciences research. During actual experiments, adjustments and optimizations to the experimental steps and analysis methods may be required based on the specific experimental objectives and cell types.