This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Before you move on to blocking, make sure the membrane is worth taking forward.
A Western blot can start failing long before the antibodies ever touch the membrane. If transfer is incomplete, uneven, or locally disrupted, you may not realize it until much later—after blocking, primary incubation, washes, secondary, and detection. By then, transfer problems are harder to separate from antibody or detection problems, and the blot has already cost you time.
That is why one of the most useful QC steps in the workflow happens immediately after transfer and before blocking.
At that point, you are not trying to finish the analysis. You are trying to answer one practical question: Is this membrane good enough to keep going—or am I about to waste the blot?
Check transfer quality right after transfer and before blocking.
For many routine blots, Ponceau S is enough for a first-pass check. It can show whether protein reached the membrane, whether lanes look broadly even, and whether there are obvious local defects.
If you need a clearer lane-level readout—or already expect total protein signal to matter later—a total protein stain may be the better choice. A loading control can still be useful later, but it should not be your first transfer QC step.

A practical guide to choosing the right membrane based on workflow, reprobing needs, and downstream use.
Western blot problems are often blamed on antibodies, transfer conditions, or blocking. But in many experiments, the first preventable mistake happens even earlier: membrane selection.
If you are deciding between PVDF and nitrocellulose for Western blot, the best choice depends on what the blot needs to do after transfer. As a practical rule, PVDF is often the better choice for stronger protein retention, reprobing, and more demanding downstream workflows, while nitrocellulose is often the more practical choice for simpler, routine Western blotting.
A membrane that fits one workflow well may be less suitable for another. The right choice affects how the blot behaves during detection, whether it holds up for reprobing, and how easy the overall workflow is to manage.
This article focuses on that bench-level decision. Rather than repeating general membrane definitions, it is designed to help you choose between PVDF and nitrocellulose based on workflow, detection needs, and downstream use.
If your workflow involves reprobing, more demanding target detection, or stronger emphasis on membrane durability, PVDF is often the stronger fit. It is commonly chosen when researchers want a membrane that can hold up well beyond a simple one-time readout.
If your blot is straightforward, single-round, and focused on day-to-day practicality, nitrocellulose is often the easier choice...

Choosing a lysis buffer is only part of protecting your sample. Inhibitor timing can determine whether your blot reflects the biology of the sample—or changes introduced during preparation...

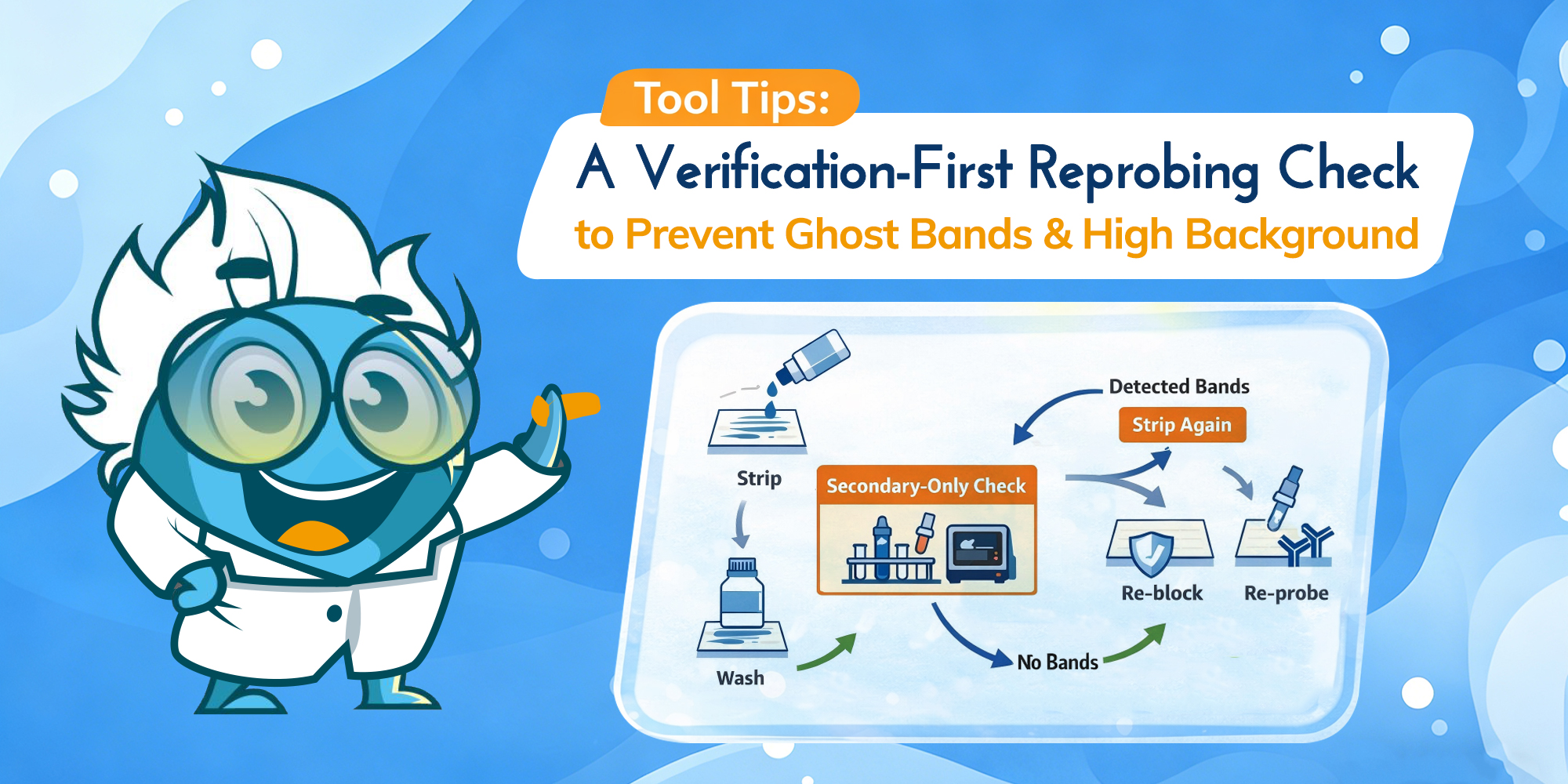
A verification-first workflow to prevent ghost bands and high background.
Western blot “failures” are often pinned on antibodies, transfer, or blocking. But when you’re stripping and re-probing, the make-or-break step is simpler: whether round one antibodies are truly removed without damaging what you’re trying to detect next. The goal is a verification-first stripping protocol that keeps signal-to-background high—so round two reads like biology, not carryover.
If you want broader context (or want to move downstream after your workflow is solid), these internal hubs are designed to be your next clicks:
Stripping and re-probing is most useful when it replaces a full rerun—another gel, another transfer, and another antibody cycle—without compromising interpretability. The workflow below focuses on the two outcomes that matter most in practice: avoiding antibody carryover that becomes ghost bands, and preserving immobilized protein so your second-round signal doesn’t collapse into background.
Stripping is worth doing when reusing the membrane genuinely replaces a full gel/transfer cycle. But it can become a time sink when your first-round signal is already near the detection limit or your experiment requires strict quantitative comparability across conditions. When you’re unsure whether your problem is stripping-related or coming from upstream steps, it’s often faster to cross-check your baseline workflow against the Western blot troubleshooting library before you change stripping conditions.
Table 1. Strip & re-probe vs rerun: a decision guide
| Situation | Strip & re-probe is usually a good idea | Rerun is usually the safer choice |
|---|---|---|
| Sample amount | Sample is limited and lanes are precious | Sample is not limiting |
| Signal strength | First-round bands are clear and usable | First-round bands are weak or near background |
| Targets | You need two targets, or phospho → total | You need many targets across many rounds |
| Data requirements | Confirmatory readout or limited reprobing | Strict quantitation with minimal added variability |
| Risk tolerance | You can accept 1–2 reprobing rounds | You can’t risk losing a low-abundance target |
A practical rule that saves time: if the band is barely above background in round one, stripping rarely “rescues” the experiment. It usually increases variability and makes the second round harder to interpret.
Quantitation note: Reprobing is best for adding a second readout or confirming changes. If you need publication-grade quantitation across multiple rounds, rerunning separate blots is typically more defensible than relying on many stripping cycles.
A stripping protocol only works when it removes antibodies without stripping away what you actually need—the immobilized protein. For that reason, the safest default is to start with milder stripping conditions and escalate only when you have evidence that antibodies remain. Many ghost band and background issues are not caused by “weak stripping,” but by incomplete removal of stripping reagents and antibody fragments during washing.
Bench note (scope): Stripping performance depends strongly on membrane type (PVDF vs nitrocellulose), detection chemistry (HRP/ECL vs fluorescence), and antibody affinity. Treat “mild vs harsh” as a range rather than a single recipe, and validate with the secondary-only check on your specific membrane + detection setup.
Table 2. Mild vs harsh western blot stripping: typical outcomes
| Approach | Best for | What can go wrong | What to adjust first |
|---|---|---|---|
| Mild stripping | Preserving signal; first attempt; sensitive targets | Residual antibodies → ghost bands | Improve wash exchanges; verify with secondary-only check; repeat stripping incrementally |
| Harsh stripping | Stubborn carryover after verification | Protein loss → weaker bands; surface stress → higher background | Shorten exposure; step down force; keep rounds limited |
The protocol below is designed to keep reprobing predictable. The key idea is that verification is part of the protocol—not an optional add-on.

Verification-first workflow. Strip gently, wash thoroughly, then use a secondary-only check before re-probing. (Click to open full-size.)
Start with the mildest stripping condition that can remove bound antibodies. If you’re unsure, avoid defaulting to long incubations. Over-stripping can reduce recoverable signal and can also make background harder to control in later rounds.
Wash thoroughly in TBST (or your standard wash buffer). What matters most is not just wash time; it’s whether you are doing full solution exchanges to remove stripping reagents and any released antibody material. If you’re standardizing your workflow for consistency, your choice of buffers, substrates, membranes, and related essentials often lives in one place—your Western blot reagents setup.
What we mean by “buffer exchange”: replace the wash buffer with fresh TBST each time under agitation, rather than extending a single wash in the same buffer.
After stripping and washing, incubate the membrane with secondary antibody only (no primary), wash, then do a short exposure. This is the fastest, most reliable way to detect residual antibody signal before you invest in another full primary incubation.
Secondary-only check (operational definition): re-block the membrane, incubate with the same secondary used in round one (same species and detection chemistry), wash under the same rules, then take a short exposure that would have detected the original band. Use your round-one exposure as a reference point; the goal is to detect residual signal without overexposing the membrane. This check is only interpretable when the secondary and detection settings match what you used previously.
Table 3. Secondary-only check: how to confirm stripping worked
| What you see | Most likely meaning | What to do next |
|---|---|---|
| Clear bands (especially at prior target MW) | Antibody carryover or incomplete stripping | Increase TBST wash exchanges; repeat stripping incrementally; re-check |
| Diffuse haze / elevated background | Residual stripping reagent or insufficient re-blocking | Wash more thoroughly; re-block longer; lower secondary concentration |
| Clean image (no bands) | Membrane is ready for reprobing | Proceed to re-blocking and primary incubation |

Secondary-only check. Use the pattern to decide whether to wash more, strip again, or proceed to re-probing. (Click to open full-size.)
Re-blocking helps stabilize membrane surface behavior after stripping. When you re-probe, start with a validated antibody dilution rather than increasing concentration to “force” signal—on post-strip membranes, aggressive antibody concentrations often increase background faster than true signal.
Reprobing order tip: probe the most sensitive/low-abundance target first (before the membrane sees repeated processing), then reprobe higher-abundance targets or loading controls later. If phospho/total is your goal, phospho is typically probed first, then strip and probe total protein.
If your second round is aimed at a loading control, plan that choice deliberately. Many workflows rely on a stable loading control as the anchor for interpretation; for options that match your species and sample type, see Loading control antibodies. If your experiment depends on rigorous normalization across conditions, it also helps to align your strategy with Total protein normalization vs loading control antibodies before you decide which readout belongs in which round.
A realistic operating range is one to two reprobing rounds. Additional rounds can work, but signal loss and background drift become increasingly likely, especially for low-abundance targets.
When reprobing fails, the symptom usually points directly to the correct lever. Ghost bands indicate antibody carryover, which is best addressed by washing and verification before escalating stripping strength. Weak second-round signal points toward over-stripping and calls for milder conditions or shorter exposure. Background haze commonly reflects residue and membrane surface effects, so washing and re-blocking dominate the fix. For pattern matching and upstream checks, the Western blot troubleshooting library is often the fastest way to identify whether you’re seeing carryover, non-specific binding, or a transfer/sample issue that stripping won’t solve.
If your experiment requires multiple targets with defensible comparability, the “one membrane, many rounds” strategy often stops being efficient. In those cases, rerunning separate blots—or outsourcing a critical target to a Western blotting service workflow—can be faster than repeated stripping iterations, especially when sample is limited or the target is low-abundance.
A western blot stripping buffer removes bound antibodies (primary and/or secondary) from the membrane so the blot can be probed again for a different target.
A reliable protocol uses the mildest stripping condition that works, thorough TBST washes with full exchanges, and a secondary-only verification step before reprobing.
Use a secondary-only check after stripping. If bands remain, improve washing first and repeat stripping incrementally before reprobing.
High background is often caused by incomplete removal of stripping reagents, insufficient re-blocking, or overly concentrated antibod...
Western blot “quantification” is often treated like a software task. In reality, most wrong numbers come from two upstream issues: saturation (signal no longer scales with protein amount) and normalization (a reference that shifts or saturates). The goal is not “the darkest band,” but measurable signal in the linear range with a defensible reference—so your fold change reflects biology, not imaging artifacts.
This post is a practical, 5-minute workflow for western blot quantification: capture a quantifiable image, measure band intensity (densitometry), normalize (loading control or total protein), and calculate fold change. If you want broader context or need to fix blot quality first, these internal hubs are designed to be your next clicks:

Western blot “failures” are often blamed on antibodies, transfer, or blocking. But in many cases, the real bottleneck happens earlier: lysis choice plus lysate handling. A buffer that’s too mild can leave your target behind. A buffer that’s too harsh can produce viscous, debris-rich lysate that smears lanes and raises background. The goal is not “the strongest lysis possible,” but the mildest system that reliably extracts your target with the best signal-to-background—and then handling it in a way that keeps lanes clean.
This post is the very beginning for our Western blot experiment starting from sample prep. If you want broader context (or want to move downstream after lysate quality is solid), these internal hubs are designed to be your next clicks:


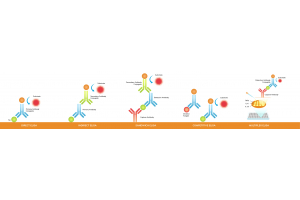
Blotting techniques are essential tools in molecular biology and biochemistry, allowing researchers to detect and analyze nucleic acids and proteins. These techniques enable scientists to uncover intricate details about genetic material, gene expression, and protein interactions. From the foundational Southern blotting, which revolutionized DNA analysis, to the versatile Western blotting, a staple in protein research, blotting methods have become indispensable tools in the laboratory.
This blog will delve into 10 blotting types, each with unique applications and methodologies. We'll explore the nuances of each technique, highlighting their roles in studying DNA, RNA, and proteins.
If you’re looking to learn about the different types of blotting techniques for your research, this guide is for you!
Southern blotting, the original blotting technique, is named after its inventor Edwin Southern who developed the method in 1975 for transferring DNA from a gel to a membrane, enabling the identification of specific DNA sequences. The naming convention for subsequent blotting techniques was influenced by this original name.
To this day, Southern blotting remains a common technique for DNA analysis and identification. The process involves digesting DNA with restriction enzymes, separating the fragments by gel electrophoresis, and transferring them onto a membrane. Labeled DNA or RNA probes then hybridize to the target sequences. Popular in genetics and molecular biology, Southern blotting is used for the detection of specific DNA sequences, study of DNA methylation patterns, RFLP (Restriction Fragment Length Polymorphism) analysis for genetic fingerprinting, identification of gene mutations and polymorphisms, and gene mapping and cloning.
Northern blotting was named by James Alwine, David Kemp, and George Stark in a 1977 paper as a playful pun on Southern blotting, indicating its use for RNA rather than DNA.
This technique is similar to Southern blotting, but focuses on RNA analysis. RNA samples are separated by gel electrophoresis, transferred to a membrane, and hybridized with labeled DNA or RNA probes to detect and quantify specific RNA transcripts. Northern blotting is commonly used to analyze gene expression patterns, determine mRNA size and abundance, study RNA processing and degradation, and detect alternative splicing events. To quantify RNA expression levels beyond blotting techniques, a solid qpcr service provides a highly sensitive and accurate alternative for transcript analysis.
Western blotting, also called immunoblotting, is widely used for protein detection and analysis, making it the most popular blotting technique. For streamlined and reproducible results, many researchers turn to a dedicated Western Blotting Service to handle the full workflow with optimized protocols. Proper western blot sample preparation is essential to ensure reliable and reproducible protein detection throughout the process. Understanding the western blot principle is key to effectively applying this method and interpreting its results accurately. The method was first described by Towbin et al. in 1979 and the term "western" was coined by W. Neal Burnette in 1981 as a tongue-in-cheek reference to the direction naming theme established by Southern and Northern blotting, this time focusing on proteins instead of nucleic acids.
This technique detects specific proteins by separating them via gel electrophoresis (typically SDS-PAGE), transferring them to a membrane (usually PVDF or nitrocellulose: AR0135-02, AR0135-04), and using primary and secondary antibodies for detection. The secondary antibody is usually conjugated to an enzyme or a fluorescent tag that produces a detectable signal when exposed to a substrate or under specific conditions. For your western blot experiment, you can explore Boster’s catalog to browse primary antibodies and secondary antibodies validated for western blot as well as find western blot reagents you will need.
Before electrophoresis begins, sample extraction quality already shapes the final blot. This guide on which lysis buffer to use for Western blot helps match buffer choice to target protein type, solubility, and downstream blot performance.
After protein extraction and electrophoresis, transfer is the next major checkpoint. This practical guide on how to check transfer quality in Western blot is useful for confirming whether weak or uneven signal comes from transfer failure rather than from the antibody step alone.
If you are deciding between PVDF and nitrocellulose for sensitivity, background, binding capacity, or handling preference, this guide on which membrane to choose for Western blot offers a more practical side-by-side comparison for real workflow decisions.
For researchers moving from detection to measurement, this guide on western blot quantification provides a practical workflow for densitometry, normalization, and fold-change calculation, with particular emphasis on staying in the linear range and avoiding saturation-related errors.
If the same membrane needs to be reprobed for another target or for loading-control confirmation, this Western blot stripping buffer protocol provides a practical starting point for removing bound antibodies while preserving usable blot signal.
In protein research and diagnostics, Western blot is used for the detection and quantification of specific proteins in a sample, analysis of protein expression levels across different conditions or treatments, detection of post-translational modifications (e.g., phosphorylation, glycosylation), study of protein-protein interactions, and confirmation of protein identity and purity in recombinant protein production. For researchers interested in analyzing protein expression directly within intact cells, consider our in-cell western blot service, which offers a high-throughput, quantitative alternative to traditional blotting techniques.
To learn more about western blotting, download our Western Blot eBook, which discusses the principle, protocol, troubleshooting tips, and FAQs for western blot.
Following the directional theme set by Southern and Western blotting, Eastern blotting is used to analyze post-translational modifications (PTMs) of proteins, such as glycosylation or phosphorylation. Many scientists deem Eastern blotting as a variation of Western blotting.
The Eastern blot technique involves transferring proteins separated by gel electrophoresis onto a membrane, followed by detection using specialized probes or antibodies targeting the PTM of interest. Though less frequently performed than other blotting techniques, Eastern blotting is useful for analyzing PTMs (e.g., glycosylation), studying protein modifications (e.g., phosphorylation, lipidation), and detecting and characterizing glycoproteins and other modified proteins.
Far-Western blotting, derived from Western blotting, focuses on protein-protein interactions. It involves transferring proteins separated by gel electrophoresis onto a membrane and identifies interactions using labeled proteins or peptides to probe for binding with the immobilized target proteins.
Growing in popularity, Far-Western blotting is valuable for identifying and studying protein-protein interactions, mapping interaction domains, and screening potential binding partners.
Southwestern blotting combines features of Southern and Western blotting techniques, focusing on DNA-binding protein detection. This technique involves transferring denatured proteins from a gel onto a membrane, followed by incubation with labeled DNA probes to identify proteins that bind to specific DNA sequences.
Though less common than Southern and Western blotting, Southwestern blotting is useful for detecting DNA-binding proteins, studying protein-DNA interactions, and identifying transcription factors and other regulatory proteins.
Reverse Northern blotting indicates the reversal of the Northern blotting process. Instead of transferring RNA and probing with DNA, Reverse Northern blotting transfers DNA and probes with labeled RNA.
This technique involves immobilizing DNA on a membrane and hybridizing it with labeled RNA or cDNA probes to detect DNA sequences. Although less common than standard Northern blotting, it is used for gene expression analysis using cDNA or genomic DNA arrays, screening differentially expressed genes in various conditions, and studying changes in transcript levels in response to treatments.
Colony blotting is named for its application in screening microbial colonies, such as bacterial or yeast colonies. It involves transferring entire colonies from a culture plate onto a membrane, where specific nucleic acids or proteins are detected through DNA hybridization analysis.
Primarily used in microbiology and cloning, colony blotting helps screen colonies for target DNA or RNA sequences, identify recombinant clones containing specific genetic inserts, and rapidly detect plasmid-containing colonies.
Dot blotting is frequently used as a quick and simple screening method for the presence or absence of nucleic acids and proteins. Named for the dot-like application of samples on the membrane, it is a simplified version of Western blotting, where samples are directly spotted onto a membrane for detection without prior gel electrophoresis.
Popular for rapid screening, dot blotting is used to screen specific nucleic acids or proteins, relatively quantify target molecules, and analyze large numbers of samples simultaneously. It should be noted that dot blots do not provide information about molecular weight, so false positive signals or the presence of modified proteins are difficult to identify.
Slot blotting is similar to dot blotting, but less commonly used. Named for the slot-like application of samples, slot blotting involves applying samples in rectangular slots on a membrane, allowing more uniform application and the quantification of target molecules in a sample without the need for gel electrophoresis.
This technique is helpful for analyzing nucleic acids or proteins without electrophoresis, comparing the relative abundance of target molecules in different samples, and screening multiple samples in high-throughput formats. As with dot blots, slot blots are also unable to provide information about the size of the target protein.
We have provided a table below that highlights the features of each blotting type.
| Blotting Type | Target Molecule | Detection Method | Technique Description | Applications |
|---|---|---|---|---|
| Southern Blotting | DNA | Labeled DNA/RNA probes | DNA fragments are separated by electrophoresis, transferred to a membrane, and probed. | Detection of DNA sequences, genetic fingerprinting, gene mapping |
| Northern Blotting | RNA | Labeled DNA/RNA probes | RNA is separated by electrophoresis, transferred to a membrane, and hybridized with probes. | Analysis of gene expression, RNA processing, alternative splicing |
| Western Blotting | Proteins | Primary/secondary antibodies | Proteins are separated by SDS-PAGE, transferred to a membrane, and detected with antibodies. | Protein detection, expression analysis, post-translational modifications |
| Eastern Blotting | Modified proteins | Specific probes/antibodies | Proteins are transferred to a membrane and probed for post-translational modifications. | Analysis of glycosylation and other post-translational modifications |
| Far-Western Blotting | Proteins (interactions) | Labeled proteins/peptides | Proteins are transferred to a membrane, probed with labeled proteins to detect interactions. | Study of protein-protein interactions, mapping interaction domains |
| Southwestern Blotting | DNA-binding proteins | Labeled DNA probes | Proteins are transferred to a membrane and probed with labeled DNA to detect binding. | Detection of DNA-binding proteins, study of protein-DNA interactions |
| Reverse Northern Blotting | DNA | Labeled RNA/cDNA probes | DNA is immobilized on a membrane and hybridized with labeled RNA/cDNA probes. | Gene expression profiling, screening for differentially expressed genes |
| Colony Blotting | DNA/RNA in colonies | Labeled probes | Microbial colonies are transferred to a membrane and probed for specific sequences. | Screening for specific sequences, identifying recombinant clones |
| Dot Blotting | Nucleic acids/proteins | Labeled probes/antibodies | Samples are spotted directly onto a membrane for rapid detection. | Rapid screening, quantification of target molecules |
| Slot Blotting | Nucleic acids/proteins | Labeled probes/antibodies | Samples are applied in slots on a membrane for quantitative analysis. | Quantitative analysis, comparing relative abundance of target molecules |
Blotting techniques are important tools in molecular biology and biochemistry. Each blotting method caters to different experimental needs, enabling detailed analysis of DNA, RNA, proteins, and their interactions. By utilizing each technique’s unique advantages and applications, researchers can deepen our understanding of molecular interactions and processes. Boster Bio supports these workflows with advanced solutions like our Recombinant Antib...
Understanding the relative abundance of target proteins and effectively normalizing data are crucial aspects of Western blot analysis. These critical aspects are deeply rooted in the western blotting principle and procedure, which outline how proteins are separated...
Western blotting (also called Protein Immunoblotting) is an analytical technique used to detect specific proteins in the given sample. A deep understanding of the western blot principle provides the essential foundation for applying this method successfully in protein detection. It uses SDS-polyacrylamide gel electrophoresis (SDS-PAGE) to separate various proteins contained in the sample. The separated proteins are then transferred or blotted onto a matrix, where they are stained with antibodies specific to the target protein. Expression details of the target proteins in the given cells or tissue homogenate can then be obtained through analyzing the location and intensity of the specific reaction. Western blotting analysis can detect target protein as low as 1 ng due to high resolution of the gel electrophoresis and strong specificity and high sensitivity of the immunoassay. This method is used in the fields of molecular biology, biochemistry, immunogenetics and other molecular biology disciplines for various experiments. Researchers who prefer standardized, high-sensitivity workflows often rely on a professional western blot service to ensure consistent and reproducible results.
Enhanced Chemiluminescence (ECL) Western Blot Substrate is a very sensitive, non-radioactive, enhanced luminol-based chemiluminescent substrate that allows for easy detection of horseradish peroxidase (HRP) on immunoblots. HRP is a common molecule conjugated to antibodies. ECL Western Blot Substrate has the capability...