This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
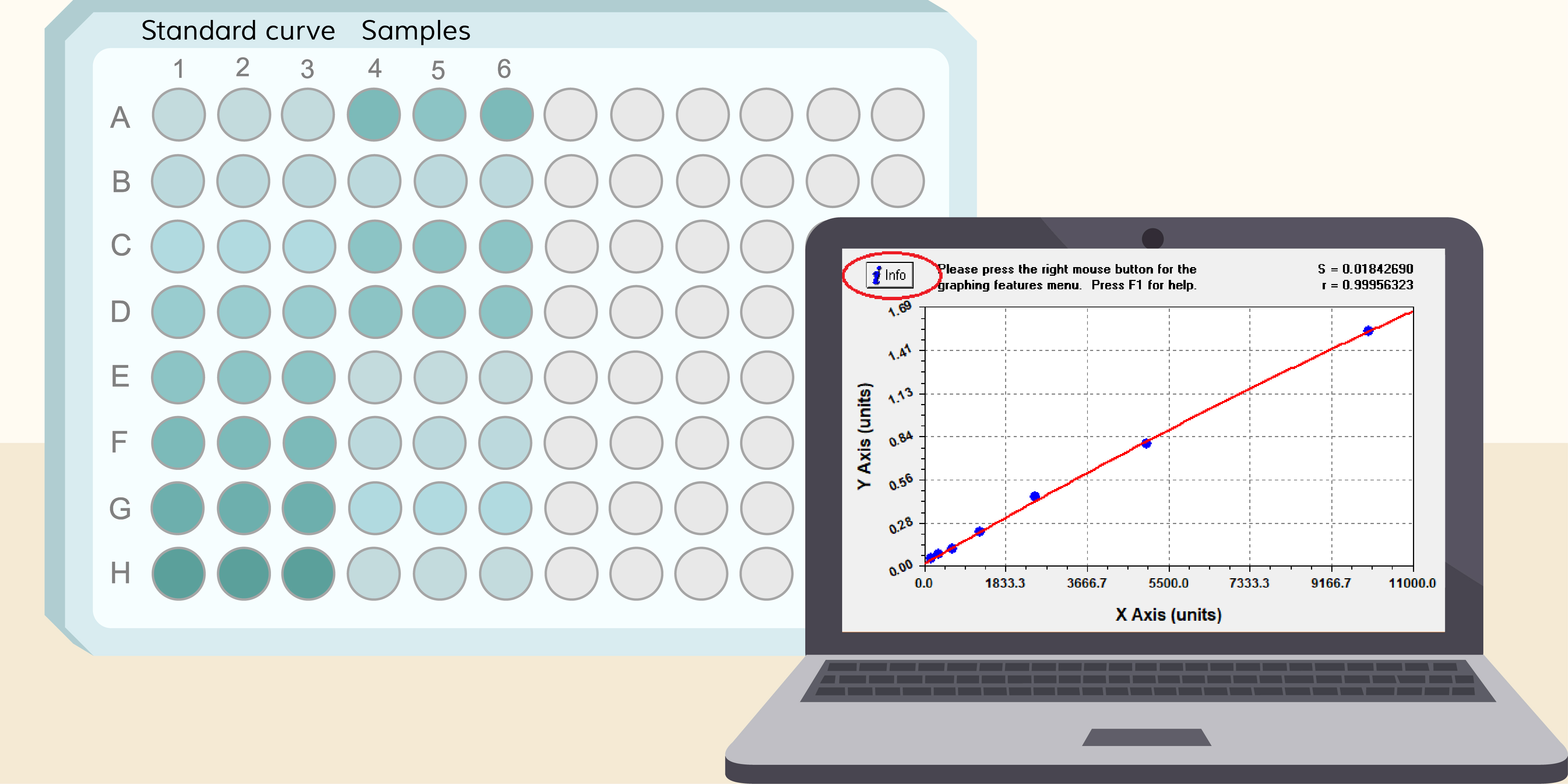
The Enzyme-Linked Immunosorbent Assay (ELISA) is a cornerstone technique in immunology and diagnostics, known for its sensitivity, specificity, and ability to detect a wide range of biomolecules. Despite its widespread use and standardized workflow, calculating and interpreting ELISA results can be complex, varying by assay type, kit manufacturer, and research purpose—each of which may follow slightly different procedural frameworks as seen in practical ELISA Testing Service implementations.
This guide provides a clear, practical overview of ELISA data analysis, from choosing the right curve-fitting model to determining when qualitative, semi-quantitative, or full quantitative interpretation is appropriate. It also includes ELISA troubleshooting tips to help ensure reliable and reproducible results. Whether you're new to ELISA or looking to improve your assay accuracy, this article will support your efforts with proven strategies and tools.
Before diving into calculation details, it can also be helpful to review an ELISA experimental design checklist so assay format, controls, standards, dilution planning, and plate reading decisions are aligned before the run begins.
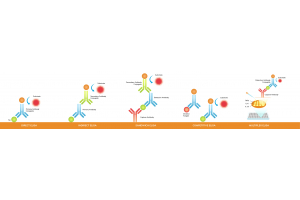
Although the core principle of ELISA is the detection of a specific molecular interaction—typically between an antigen and an antibody—there are multiple assay formats, each with distinct detection mechanisms. These differences directly influence how results should be calculated and interpreted.
The most common ELISA types include:
If you are still deciding which assay structure best fits your analyte, sensitivity target, and downstream readout, this guide on which ELISA is for you is a useful companion before locking in the analysis strategy.
Understanding your ELISA format is the first step in choosing the right data analysis strategy.
At the core of ELISA result interpretation lies the relationship between optical density (OD) and target concentration. This relationship varies depending on the ELISA format:
ELISA kits from different manufacturers may recommend different methods for fitting the standard curve, depending on the assay range and expected sensitivity:
For a more detailed walkthrough of standard preparation, concentration gradients, and graph construction, see how to generate an ELISA standard curve.
While researchers can manually fit curves using tools like Microsoft Excel or GraphPad Prism, there are also excellent online platforms that simplify the process:
Qualitative interpretation (positive vs negative) is suitable when the goal is to detect the presence or absence of a target, rather than its exact concentration. Typical scenarios include:
To determine whether a sample is positive or negative, a cutoff OD value must be established. Common methods include:
If you need a more practical breakdown of what each control is actually telling you, including process blank, true negative, and spike-based checks, this guide on ELISA controls that actually matter can help tighten interpretation.
Interpretation:
This method offers a simple yet effective way to evaluate results in screening or diagnostic assays.
In some studies, particularly when monitoring immune responses or comparing relative expression, semi-quantitative grading is helpful. Rather than reporting precise concentrations, samples are categorized by signal strength:
This approach is commonly used in:
Quantitative ELISA allows researchers to determine the absolute concentration of a target analyte in unknown samples by comparing their optical density (OD) values to a standard curve generated from known concentrations. This section provides a step-by-step guide to ensure accurate and reproducible quantification.
If you want a dedicated walkthrough for concentration gradients, blank setup, and standard-point arrangement, refer to how to generate an ELISA standard curve.
Accurate fitting of the standard curve is essential for precise interpolation of sample concentrations.
Equation form:
The 4-parameter logistic (4PL) equation is:
Y = D + (A - D) / (1 + (X / C)B)
where A = minimum, D = maximum, C = inflection point (EC50), B = slope
Once the standard curve is fitted:
For a more practical workflow on choosing starting dilution, running a pilot serial dilution, and avoiding saturation or out-of-range readings, see how to decide ELISA dilution ratio.
Ensuring accuracy and reproducibility is critical in quantitative ELISA analysis.
When reproducibility problems persist despite a smooth curve, it is often useful to step back and review controls, dilution logic, and plate setup together using an ELISA experimental design checklist and a more focused control guide such as ELISA controls that actually matter.
Even with well-validated ELISA kits and protocols, variability and errors can arise during the experiment. This section outlines frequent issues, explains their causes, and provides practical solutions.
Possible causes:
Solutions:
Possible causes:
Solutions:
Possible causes:
Solutions:
Possible causes:
Solutions:
Answer: Yes, it is strongly recommended.
Even slight changes in lab conditions (e.g., pipetting technique, plate washing, ambient temperature) can affect the standard curve. Relying on a previous curve may result in significant errors in sample concentration calculations.
| Problem | Possible Cause | Suggested Solution |
|---|---|---|
| OD too low or flat | Missing reagents, substrate expired, low concentrations, under-incubation | Check reagent setup, increase incubation, optimize concentrations |
| OD too high or background noisy | Insufficient washing, too much detection Ab, over-incubation | Wash more, dilute antibodies, optimize blocking/incubation |
| Bad standard curve fit | Pipetting error, not enough points, non-sigmoidal range | Use 4PL fitting, increase standards, check dilution accuracy |
| High CV% between replicates | Pipetting inconsistency, sample inhomogeneity, cross-contamination | Standardize pipetting, vortex samples, avoid edge wells, peel seals carefully |
| “Can I reuse old standard curve?” | Environmental and technical variability | No—always regenerate fresh standard curves per run |
| CV% still too high | Scraping coating, poor sample quality, edge effects | Avoid touching wells, pre-mix samples, use central wells only |
Accurate calculation of ELISA results is essential for obtaining reliable, reproducible data that can support meaningful biological conclusions. From understanding the type of ELISA used, to properly reading OD values and generating a high-quality standard curve, each step of the process requires careful attention. Determining whether your experiment calls for qualitative, semi-quantitative, or full quantitative analysis helps guide the appropriate interpretation strategy. Tools such as 4PL fitting and online ELISA analysis platforms—including Boster’s own analysis tool—can significantly streamline this workflow. Finally, being aware of common ELISA pitfalls and implementing best practices in pipetting, plate handling, and data analysis will minimize error and enhance your assay’s consistency. By following the methods outlined in this guide, researchers can improve the accuracy of their ELISA results and ensure greater confidence in their experimental findings.