This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
and ELISA kits, proteins related to Cystic Fibrosis.
Cystic Fibrosis (CF) is a serious genetic disorder that primarily affects the lungs and digestive system. Caused by mutations in the CFTR gene, CF leads to the production of thick, sticky mucus that clogs airways and traps harmful bacteria, resulting in chronic respiratory infections and inflammation. This persistent lung damage can severely impair breathing and reduce quality of life. Additionally, the thick mucus obstructs the pancreas, hindering the release of essential enzymes needed for digestion and nutrient absorption. Ongoing research into CF is exploring innovative treatments, including the development of targeted antibodies, to address the underlying molecular causes of the disease. These advancements aim to improve patient outcomes, enhance lung function, and extend the lifespan of those living with Cystic Fibrosis, offering renewed hope for effective management and eventual cures.
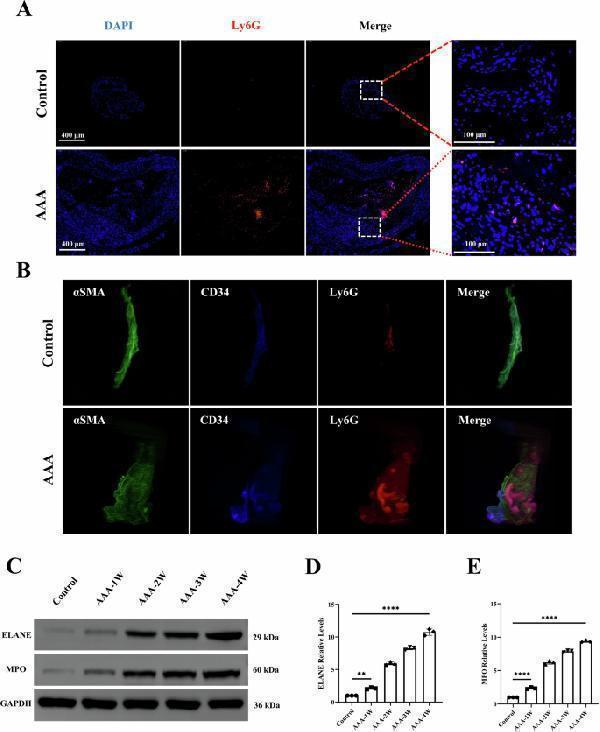
PB10058
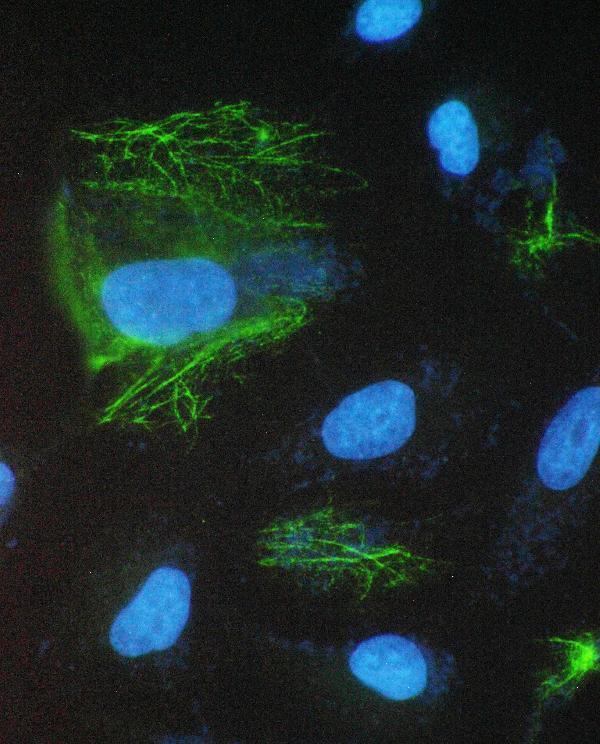
A01413-1
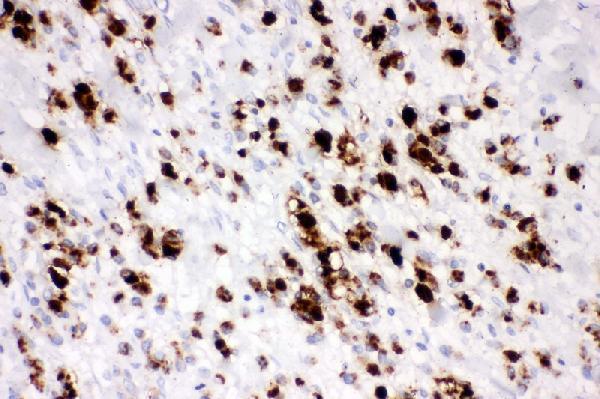
PA2303
| Protein Name | Gene Name | Function |
|---|---|---|
| Cystic Fibrosis Transmembrane Conductance Regulator | CFTR | Chloride channel for ion transport in epithelial cells |
| Epithelial Sodium Channel α | SCNN1A | Regulates sodium absorption and fluid balance |
| Epithelial Sodium Channel β | SCNN1B | Functions with ENaCα in sodium transport |
| Epithelial Sodium Channel γ | SCNN1G | Works with ENaCα and ENaCβ in sodium regulation |
| Mucin 5AC | MUC5AC | Mucin protein involved in mucus production |
| Mucin 5B | MUC5B | Mucin protein essential for mucus structure |
| Interleukin-6 | IL6 | Pro-inflammatory cytokine involved in immune responses |
| Neutrophil Elastase | ELANE | Protease from neutrophils causing tissue damage |
| Tumor Necrosis Factor-alpha | TNF | Pro-inflammatory cytokine mediating inflammation |
| Transforming Growth Factor-beta | TGFB1 | Involved in tissue fibrosis and remodeling |
| SLC26A9 | SLC26A9 | Chloride channel implicated in ion transport |
| Interleukin-1beta | IL1B | Pro-inflammatory cytokine involved in immune response |
| Interleukin-17 | IL17A | Pro-inflammatory cytokine involved in neutrophil recruitment |
| Macrophage Inflammatory Protein-1alpha | CCL3 | Chemokine involved in recruiting immune cells |
| Kallikrein-related Peptidase 5 | KLK5 | Protease involved in skin barrier function and inflammation |
| Vascular Endothelial Growth Factor A | VEGFA | Regulates vascular permeability and angiogenesis |
| Galectin-3 | LGALS3 | Involved in cell adhesion and immune response |
| Interleukin-13 | IL13 | Pro-inflammatory cytokine involved in airway inflammation |
Cystic Fibrosis (CF) is primarily caused by mutations in the CFTR gene, which encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein. This protein functions as a chloride channel, regulating the movement of ions and water across epithelial cell membranes in organs such as the lungs, pancreas, and intestines. Understanding the structure and function of CFTR is crucial for elucidating how specific mutations disrupt its normal activity, leading to the thick mucus characteristic of CF. Research in this subarea focuses on identifying the molecular mechanisms by which different CFTR mutations impair protein folding, trafficking, and channel function. Additionally, scientists are developing and optimizing therapeutic agents known as modulators—such as correctors and potentiators—that can enhance the proper folding and activity of the defective CFTR protein. These advancements not only improve our fundamental knowledge of CF pathophysiology but also pave the way for personalized medicine approaches, allowing treatments to be tailored to the specific genetic variants present in individual patients.
Chronic lung inflammation is a central feature of Cystic Fibrosis, leading to progressive respiratory decline and reduced quality of life. This subarea of research delves into the inflammatory processes that exacerbate lung damage in CF patients. Studies focus on the role of immune cells, such as neutrophils and macrophages, and the release of pro-inflammatory cytokines and chemokines that perpetuate a cycle of inflammation and infection. Understanding these pathways is essential for identifying potential therapeutic targets to mitigate excessive inflammation without compromising the necessary immune response to pathogens like Pseudomonas aeruginosa. Researchers are also investigating the interplay between microbial colonization and the host immune system, aiming to develop strategies that can effectively manage chronic infections while controlling inflammatory responses. By targeting the underlying mechanisms of inflammation, this research seeks to preserve lung function, slow disease progression, and enhance the overall respiratory health of individuals living with Cystic Fibrosis.