This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Learn the fundamentals of molecular biology, cDNA libraries and genomic libraries, and polymerase chain reaction (PCR).
Browse Boster Featured ProductsOn this page, we're laying out the fundamental principles of molecular biology, molecular cloning and library construction, and polymerase chain reaction (PCR). Refresh your understanding of molecular biology from the structure of DNA and RNA to cellular transcription. We'll also discuss cDNA libraries and genomic libraries, and explain how DNA can be replicated within a controlled experiment. Finally, you'll learn about PCR and its variations, including the basics of gel electrophoresis, data acquisition, and analysis.
Molecular biology has been the basis for the understanding of each individual step in the biology central dogma: DNA replication, DNA transcription into RNA, and RNA translation into proteins. These molecules are responsible for giving information to cells of each organism on how to survive and reproduce according to the environmental conditions at each exact moment. All this information is stored in the genetic material of cells and transferred to progeny.
Molecular biology has been the basis for the understanding of each individual step in the biology central dogma: DNA replication, DNA transcription into RNA, and RNA translation into proteins. These molecules are responsible for giving information to cells of each organism on how to survive and reproduce according to the environmental conditions at each exact moment. All this information is stored in the genetic material of cells and transferred to progeny.
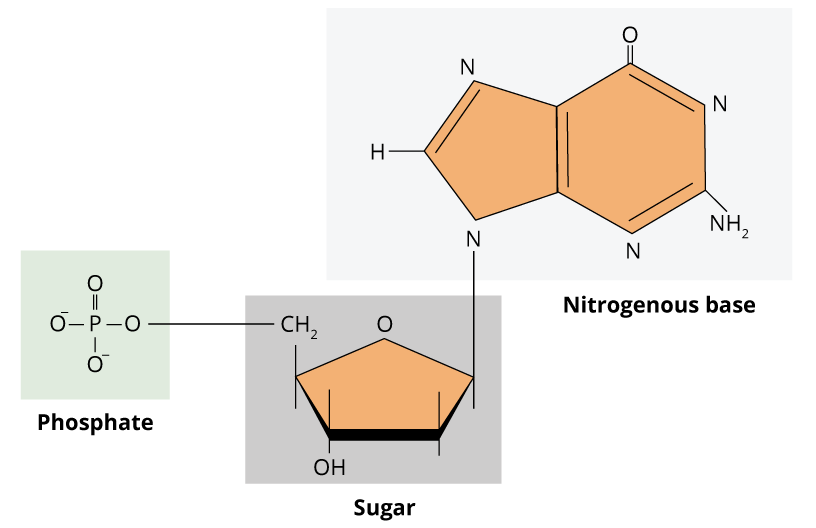
The genetic information in cells is present in nucleic acids, such as DNA and RNA. The deoxyribonucleic acid (DNA) carries the genetic code of a single cell, whereas the ribonucleic acid (RNA) is a molecule that converts this information into amino acid sequences of proteins. Genetic information relies on the sequence of monomers of nucleic acids. Thus, unlike polysaccharides and lipids that are normally formed by long repeated unities, nucleic acids are informational macromolecules. These monomers are known as nucleotides, and consequently, DNA and RNA are polynucleotides. A nucleotide is composed of three components: one pentose (ribose for RNA and deoxyribose for DNA), one nitrogenated base and one phosphate group. The structure of DNA and RNA nucleotides are very similar. The nitrogenated bases are purines (adenine and guanine, which contain two heterocyclic rings) or pyrimidines (thymine, cytosine and uracil, which contain one single heterocyclic ring).
Guanine, adenine, and cytosine are in the composition of DNA and RNA. Excluding few exceptions, thymine is present only in DNA and uracil is present only in RNA. The nitrogenated base is linked to sugar pentose by a glycosidic bond between the carbon atom of the sugar and the nitrogen atom from the base. When nitrogenated base is linked to a sugar, it is denominated nucleoside. For that reason, nucleotides, are nucleosides with an addition of one or more phosphate groups.

This guide will teach you everything you need to become a PCR expert, including a critical review of PCR and molecular biology principles, protocols, all-in-one FAQs, and more
In 1953, based on results from x-ray diffraction studies done by Williams and Franklin, the scientists Watson and Crick proposed a structural model for DNA. This model embraces both chemical and biological DNA properties, namely the replication capacity of this molecule. According to this model, the DNA molecule is formed by two helicoidal strands linked by hydrogen bonds between the nitrogenated bases of each strand. When the nitrogenated bases face each other, hydrogen bonds are formed. These hydrogen bonds are more stable between adenine and thymine, and guanine and cytosine. Thus, the adenine of one strand pairs with a thymine of the other strand and the same for guanine and cytosine. The resultant double helix strands are antiparallel, which means that the inter bonds 3′-5′ phosphodiester have opposite directions. Each round is composed by 10.5 pairs of nucleotides and measures 3.4 mm.
The size of a DNA molecule is defined as the number of nucleotide base pairs. Thus, a DNA molecule with 1000 nucleotides contains 1 kilobase (Kb). If the DNA molecule is in double helix structure, we use the base pairs nucleotide nomenclature. For instance, the bacteria Escherichia coli has around 4640 Kbp of DNA in its chromosome. Each base pair has 0.34 nanometers of length along the double helix, and each round is about 10 bp, which means that 1 Kbp of DNA represents 100 rounds, measuring 0.34 µm of length. The E. coli genome has 4640 bp and is 1.58 mm in total length. Since E. coli cells are only approximately 2 µm, the chromosome is much larger than its own cell size. For that reason, DNA needs to be compressed and packed to fit inside the cell.
The main genetic elements in cells are the chromosomes, but there are also other genetic elements such as viral genomes, plasmids, organelle genomes, and transposable elements. In prokaryotes, usually there is a single circular chromosome, while in the eukaryotes, genomes are organized in several chromosomes. Plasmids are genetic elements which replicate independently from the cell chromosomes. Usually plasmids are composed of double helix DNA molecules (circular or linear).
Transposable elements or jumping genes are DNA segments that are able to move from one site of a DNA molecule to another site of the same molecule or another distinct DNA molecule. Transposons are not found as individual DNA molecules, instead these elements are found inserted in other DNA molecules, such as chromosomes, plasmids, and viral genomes. Transposable elements are present in both eukaryotes and prokaryotes, and play an important role in genetic variation.
The gene is the basic and functional unit of genetic information. Genes are present in chromosomes or other big molecules, also referred to as genetic elements. In modern biology, the classification of organisms is made according to their genetic material composition and variability.
When genes are expressed, the genetic information stored in DNA is transferred to RNA. There are different RNA types, but only three cooperate for protein synthesis. The messenger RNA (mRNA) is a single stranded molecule that carries genetic information from the DNA to the ribosome, which is responsible for protein synthesis. The transference RNA (tRNA) convert the genetic information of RNA nucleotides into amino acid sequences of proteins. The ribosomal RNA (rRNA) is an important catalytic and structural component of ribosomes.
The molecular processes of genetic information can be divided into three stages:
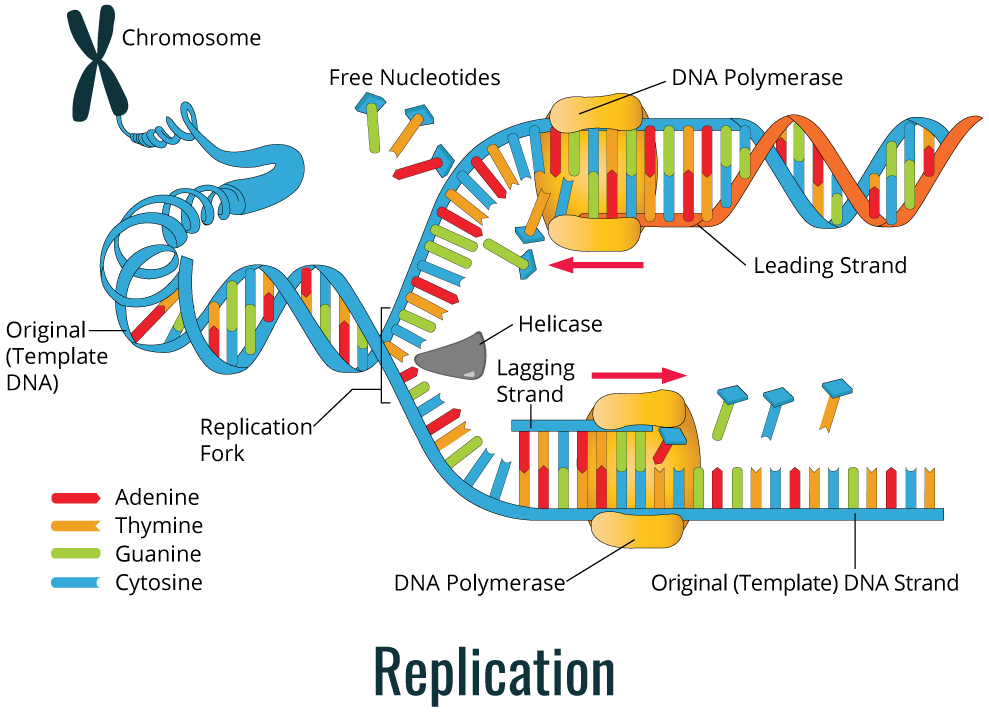
As shown in the image below, during replication, the DNA double helix is duplicated through the action of the DNA polymerase enzyme.
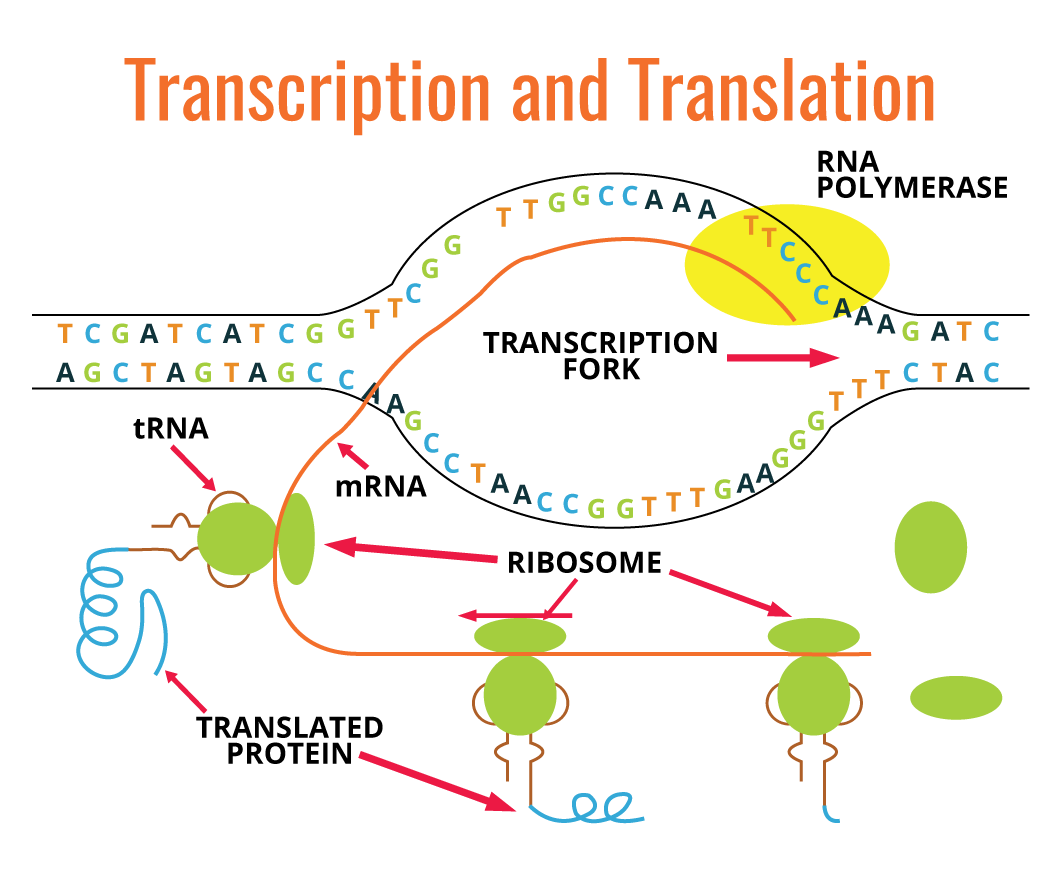
Many molecules different from RNA are transcribed from relatively short regions of DNA molecule. In eukaryotes, each gene is transcribed, generating mRNA, whereas in prokaryotes one single mRNA molecule can carry genetic information from several genes. There is a linear correlation between the nucleotide sequence of one gene and the amino acid sequence of a polypeptide. Each group of three nucleotides present in mRNA codes for one single amino acid, and each nucleotide triplet is referred to as a codon. Codons are translated into amino acid sequences by ribosomes, tRNA, auxiliary proteins, and translation factors.
The diagram above (on the right) is a simple graphic depiction of the transcription and translation processes:
Below, we will go into more detail about these processes and briefly mention their differences.
DNA replication is used by cells to allow cell division, either in reproduction or in the duplication of new cells in multicellular or unicellular organisms. The complexity of DNA replication process requires the involvement of a great number of specific enzymes. DNA is present in cells as a double helix molecule and when this helix is unwinding, a newly synthesized strand emerges along with a parental strand. The DNA strand used to produce the complementary strand is referred to as the template strand, which is used to synthesize complementary strands of each parental strand. Thus, replication is a semi-conservative process.
The diagram below (left) shows a simple color coded depiction of this semi-conservative process.

The precursor of each new nucleotide in the DNA strand corresponds to a deoxynucleoside 5’-triphosphate. During replication, two terminal phosphates are removed and the internal phosphate is covalently bonded to the deoxyribose of the raising DNA strand. The nucleotide addition requires the presence of a free hydroxyl group, which is available only at the correct end, so the addition of the nucleotide phosphate group bonds with the 3’-hydroxyl(OH) of the previous nucleotide. The enzymes that catalyze the addition of deoxy ribonucleotides are denominated DNA polymerases. There are several types of these enzymes, each one with a specific role. All known DNA polymerases work in 5’ to 3’ direction, but none of them are able to start DNA synthesis alone. Since DNA polymerase can only add nucleotides to the 3’-OH, in order to start a new strand, it requires a primer.
The primer is a nucleic acid molecule in which the DNA polymerase can add a nucleotide to. Often, the primer is a small RNA fragment instead of DNA. When the double helix is unwinding at the beginning of replication, an enzyme of RNA polymerization (primase) synthesizes the RNA primer with 11-12 nucleotides, which is complementary to the DNA template strand. At the end of the RNA primer, there is a 3’-OH group in which the DNA polymerase adds the first deoxy ribonucleotide. Later, the RNA primer will be removed and replaced by DNA.
Before DNA polymerase synthesizes a new DNA strand, the existent DNA double helix needs to undergo an unwinding process to expose the template strand. The unpackaged region is where replication will start, and it is designated the replication fork. The DNA helicase enzyme is responsible unwinding and separating the DNA double helix strands in an ATP-dependent process, exposing a small region of a single strand. Helicase can move along the double helix structure right at the front of the replication fork. There are specific regions for replication to get started, also known as replication origins.
The replication process always happens in 5’ to 3’ direction, which means that a new nucleotide is added to the 3’-OH group of the raising DNA strand. For this reason, the strand being synthesized uses the 3’-5’ strand as a template, and we call it the leading strand (continuous strand). DNA synthesis occurs continuously since there is always a free 3’-OH group. On the other hand, in the newly synthesized strand using as template the 5’-3’ DNA strand, the DNA synthesis occurs in a discontinuous process. It does not have available a free 3’-OH for a nucleotide to be added. Thus, in the lagging strand, it is necessary for primase to synthesize the primer repeatedly to make available a free 3’-OH group. In the continuous strand only one primer is required at the beginning of DNA synthesis, but the lagging strand is synthesized in short fragments, also known as Okazaki fragments. These fragments are posteriorly fused, generating a continuous strand.
A complex of proteins including DNA polymerase attaches to the DNA strand at the replication fork, and slides along the DNA template strand. Two DNA polymerases and protein complexes are required for DNA replication (one for each strand) at the replication fork. After the synthesis of the leading strand and lagging strand, a DNA polymerase with exonuclease activity is necessary to remove the RNA primer and add complementary DNA nucleotides. The last phosphodiester ligation is done by a DNA ligase enzyme. Ligase enzyme joins the DNA cuts that contain a 5’-PO4 and an adjacent 3’-OH group.
Transcription is the synthesis of ribonucleic acid RNA using DNA as template. There are three significant chemical differences between RNA and DNA:
The replacement of deoxyribose by ribose affects the chemical properties of the nucleic acid and generally enzymes that catalyze reactions in DNA do not have any action in RNA (and vice-versa). Meanwhile, the substitution of thymine by uracil does not affect the base pairing, since both thymine and uracil pair with adenine with the same efficiency.
All RNA molecules are the product of DNA transcription. They play a role at two different levels, genetic and functional. At the genetic level, the mRNA carries the genetic information from the genome to the ribosome. On the other hand, the rRNA play a functional and structural role in the ribosomes and the tRNA are responsible for transporting the amino acids for protein synthesis. Some RNA molecules, including rRNA, can have enzymatic activity.
The transcription of genetic information is done by an RNA-polymerase enzyme in a similar fashion as DNA-polymerase does in DNA replication. RNA polymerase catalyzes the formation of phosphodiester between ribonucleotides. This is driven at the expense of energy released by hydrolysis of two phosphate bonds from ribonucleotides. Similarly to DNA synthesis, RNA synthesis is performed in the 5’-3’ direction, with ribonucleotides being added to a free 3’-OH group from a previous ribonucleotide.
Unlike DNA polymerase, RNA polymerase is able to start new strands independently. Consequently, there is no need for a primer. In order to start RNA synthesis, it is necessary that RNA polymerase recognizes the DNA initiation sequences, also referred to as promoters. After RNA polymerase binds the promoter, transcription is allowed to start. In this process, the DNA double helix at the promoter region is unwound by RNA polymerase, exposing the DNA template strand. When a DNA region has two close promoters, with opposite directions, transcription takes place using both DNA strands as templates but in different directions. When the newly synthesized RNA strand dissociates from DNA, the unwinding DNA closes again back to its original double helix structure.
RNA polymerase uses DNA double strand (dsDNA) as template, however, only one of the strands is transcribed for each gene. Compared to DNA replication where all genomic DNA is replicated, in transcription only small DNA fragments are transcribed. Usually, these fragments correspond to one gene. This system allows the cell the possibility to transcribe different genes, at distinct frequencies depending on the cell's requirements.
Usually, the transcription process involves the transcription of genes required for the cell at that exact time, which means that it is critical that transcription is finished at the right spot. This termination process is controlled by specific sequences of nucleotides in the DNA template strand.
Unlike bacteria, a great number of transcripts from eukaryotes have introns (unnecessary regions for translation), which will require further RNA processing. This RNA processing is called splicing and occurs in the cell’s nucleus. The splicing involves a protein complex, the spliceosome, which is responsible for removing the introns from transcripts and joining the remaining sequences, referred to as exons.
Additionally, there are two other steps for mRNA processing in Eukaryotes. The first one is the capping process, which occurs before transcription termination. The capping process is the addition of a methylated guanine nucleotide cap at the 5’-end of the pre-mRNA. This cap will be crucial for translation initiation. The second step includes cleavage of transcript 3’-end, followed by addition of 100-200 adenylate residues, in a poly-A tail. This tail gives stability to the mRNA and its degradation will be required to allow RNA degradation.

You can access more information about sample preparation, protocols, and troubleshooting in our PCR Technical Resource Center.
Molecular biology has long been the basis for the understanding of each individual step in the biology central dogma: DNA replication, DNA transcription into RNA, and RNA translation into proteins. These molecules are responsible for giving information to cells of each organism on how to survive and reproduce according to the environmental conditions at each exact moment. All this information is stored in the genetic material of cells and transferred to progeny as well.
Manipulation and investigation of this genetic material are done through various techniques including traditional molecular cloning (library construction), Polymerase Chain Reaction (PCR), qRT-PCR, and much more. These molecular biology techniques have various broad and useful applications in our scientific community including:

This comprehensive manual will guide you through the basic principles and techniques relevant to molecular biology research, including protocols and troubleshooting solutions. Download this free eBook for a refresher course on the fundamental principles of molecular biology and PCR.
Download PCR eBookMolecular cloning is one of the most fundamental techniques of molecular biology used to study protein function and structure. Generally, in this technique, DNA coding for a protein of interest is cloned using PCR and/or restriction enzymes into a plasmid. All engineered plasmids or expression vectors have 3 main distinctive features:
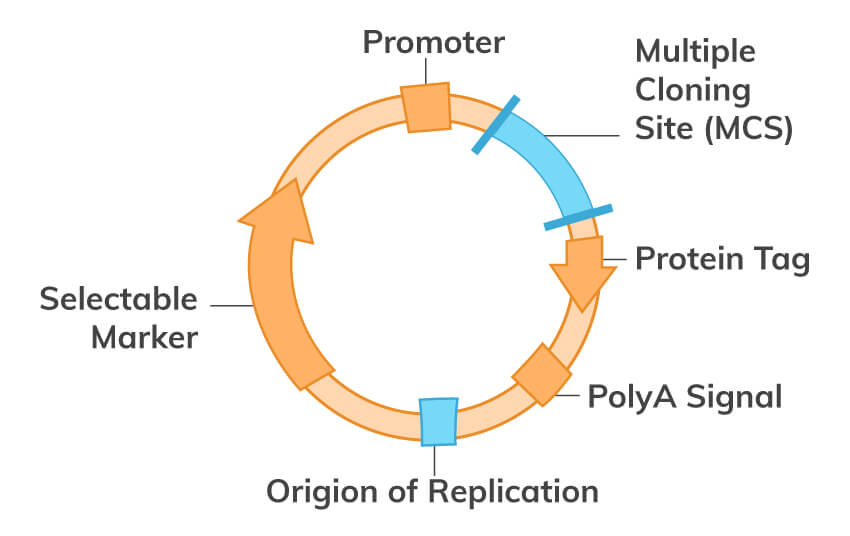
In the wild, a certain plasmid can be introduced into prokaryotic cells by transformation via uptake of naked DNA, by conjugation via cell-cell contact or by transduction via viral vector. On the other hand, the plasmid can also be inserted artificially into prokaryotic and eukaryotic cells, such as fungi and plant cells or complex animal cells. Introducing DNA into cells can be achieved by physical or chemical means, and is called transformation. Some available transformation techniques include electroporation, microinjection, calcium phosphate transfection, and liposome transfection. A stable transformation may result, wherein the plasmid is integrated into the genome. Or if the plasmid remains independent of the genome, we have what is called transient transfection. For transient transfections, since the DNA introduced is usually not integrated into the nuclear genome, the foreign DNA will be diluted through mitosis or degraded over time
In a more stable transformation, the transfected gene can be inserted in the genome of the cell, which guarantees its replication in the cell’s progeny. Usually, a marker gene is co-transformed with the gene of interest, which gives the transformed cell some selectable advantage, such as antibiotic resistance. Some of the transformed cells will (by random chance) have integrated the foreign genetic material. When antibiotic is added to the cell culture, only those few cells with the marker gene integrated will be able to survive and proliferate, while other cells die. This is the process of screening. After applying this selective stress, over time, only the cells with a stable transfection survive and can be selected. Some examples of common agents for selecting stable transfection include; Ampicillin, Kanamycin, Zeocin, Puromycin, Blasticidin S, Hygromycin B, and G418, which is neutralized by the product of the neomycin resistance gene.
The protein of interest can now be expressed by the cell using the genetic information encoded in the inserted DNA. A variety of systems are readily available to help express this protein at high levels. This protein can then be tested for enzymatic activity under different situations, studied in the pharmaceutical industry, and/or the protein may be crystallized to study its tertiary structure. The possibilities are endless with molecular biology!
DNA library technology is a fundamental technique of current molecular biology, and the range of applications of these libraries vary depending on the source of the original DNA fragments. Molecular cloning techniques allow scientists to create and store a group of DNA fragments from different sources in a suitable microorganism and take advantage of the cell machinery to protect and replicate these exogenous DNA fragments. In molecular biology, this procedure is known as DNA library construction, and we can store different kinds of genetic material, including; cDNA (formed from reverse-transcribed RNA), genomic libraries formed from genomic DNA, allele mutants, mitochondrial DNA, randomized mutant libraries, and more.
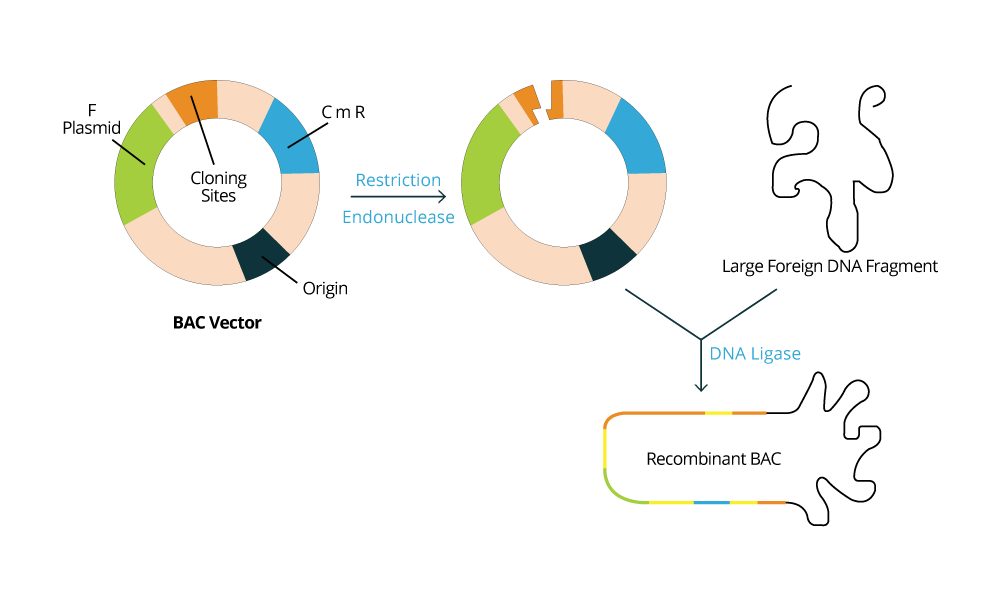
Usually, the DNA fragments intended to be stored are inserted into cloning vectors or plasmids, and the type of vector to be used will depend on the host microorganism that is used as the biological library. For example, if the microorganism selected is a bacteria like E. coli, it is expected for one to use a BAC vector (Bacterial Artificial Chromosome vector). Whereas if a yeast such as S. cerevisiae is selected to store DNA fragments, then a YAC vector (Yeast Artificial vector) is used. The host cell machinery will be responsible for maintaining and replicating the exogenous DNA fragments according to the information contained in the vectors used. The population of bacteria or yeast will be constructed such that each organism contains on average one construct (vector + insert). As the population grows in culture, the DNA molecules contained within them are "cloned" (copied and propagated).
In the left image, we have an example of a BAC vector which is commonly used.
| Vector | Properties | Size (kb) |
|---|---|---|
| BAC (Bacterial Artificial Chromosome) | Bacteria F factor and origin of replication | 75 - 300 |
| YAC (Yeast Artificial Chromosome) | Yeast centromere, and telomere | 100 - 1000 |
| MAC (Mammalian Artificial Chromosome) | Mammalian centromere and telomere and origin of replication | 100 - >1000 |
| Plasmid | Multicopy plasmids | 10 |
| Phage | Bacteriophage Υ | 5-10 |
| Cosmid | Bacteriophage Υ cos site | 35-35 |
This section goes over the typical experimental setup for cDNA library construction and basic principles. The cDNA library uses mRNA as the source of information, so it only includes the organism’s expressed genes from a particular source. This mRNA can be extracted from the organism’s cells, a specific tissue, or even an entire organism. mRNA needs to be converted into cDNA by reverse transcriptase, in order to allow the host organism to perform the correct replication and transcription processes for mRNA. This means that it represents the genes that were being actively transcribed in that particular source under the exact conditions (physiological, environmental and developmental) that existed at the time the mRNA was extracted and purified. cDNA libraries are useful in reverse genetics, although they only represent less than 1% of the overall genome in a given organism
The main advantage behind the choice of mRNA as the source for the DNA library preparation is to avoid junk DNA from genomic DNA and introns from eukaryotic genes. This ensures that only expressed genes from a specific cell or tissue are being stored. Although bacterial cells do not process introns, the use of mature mRNA (which is already spliced) allows the successful expression of the cDNA genes in bacterial host cells for further physiological tests.

cDNA libraries are used to express eukaryotic genes in prokaryotes since it does not include introns, and therefore, can be expressed in prokaryotic cells. cDNA libraries remove the large numbers of non-coding regions from the library, and it is also useful for subsequently isolating the gene that codes for that mRNA.
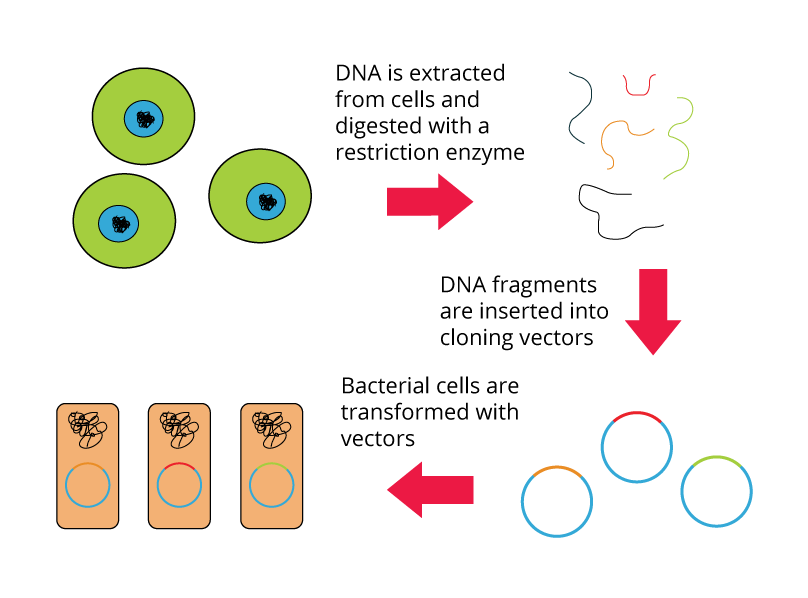
A genomic library is a collection of clones that together represent the total genomic DNA from an organism of interest. The DNA is stored in a population of identical vectors, each containing a different insert of DNA. The number of clones that make up a genomic library depends on:
The tissue source of the genomic DNA is usually not important because each cell contains the same DNA (with few exceptions). To construct a genomic library of a specific organism, several variables should be taken into account, such as: host organism, type of vector, cell transcription and translation machinery, etc. However, the most fundamental criteria is to use a host prokaryotic organism if the genome to be collected is from a prokaryotic organism, and the same principle if the genome is extracted from a eukaryotic organism (which should be stored in eukaryotic host). This selection criteria will be critical to increase the chances of successful heterologous protein expression and allow further physiological tests.
This procedure allows us to store the entire genome of a specific organism by inserting it in a specific vector with different fragment sizes depending on the restriction enzymes used. The vectors carrying small pieces of the genome are then transformed in host organism cells for further laboratory analysis.
Shown below are the steps for creating a genomic library from a large genome:
As an example, prokaryotic organisms from soil are usually targeted for genomic library procedures, largely because they are known for their ability to be resistant to several antibiotics. In vitro culture of these organisms is rather difficult, which impairs physiological tests to identify genes involved in antibiotic resistance.
The typical flowchart protocol for DNA library construction (described in previous image) should be followed. First extract genome from soil organisms, digest the genome, insert into an appropriate vector, and transform in a prokaryotic organism (e.g. E. coli). Then, E. coli transformants should be plated in LB culture medium containing a specific antibiotic, like kanamycin. Only the E. coli transformants that carry genes from soil organisms along with antibiotic resistance marker will be able to grow. Positive colonies should be selected to sequence the vector and identify genes that play a role in antibiotic resistance.
Download troubleshooting handbooks for IHC, Western blot, ELISA, PCR, and more for FREE.
How PCR works and the differences between PCR assays
Polymerase chain reaction (PCR) is a technique that widely used in molecular biology and genetics that permits the analysis of any sequence of DNA or RNA. PCR allows a specifically targeted DNA sequence to be copied and/or modified in predetermined ways. This reaction has the potential to amplify one DNA molecule to become over 1 billion molecules in less than 2 hours. This powerful and versatile technique can be used to introduce restriction enzyme sites to ends of DNA molecules, or to mutate particular bases of DNA (a technique called site-directed mutagenesis). PCR is also useful for determining whether a particular DNA fragment is found in a cDNA library. Today, PCR has been further developed to include many variations, like reverse transcription PCR (RT-PCR) for amplification of RNA, and quantitative PCR which allows for quantitative measurement of DNA or RNA molecules
For different molecular procedures, several copies of a specific DNA sequence are required. Quantitative PCR methods allow scientists to estimate the amount of a given sequence present in a sample. This technique is often applied to quantitatively determine levels of gene expression, and is an established tool that measures the accumulation of DNA product after each round of PCR amplification.
Molecular biologists realized that mimicking the cell mechanisms of DNA replication, they would be able to replicate DNA sequences of interest. In 1983, Kary Mullis developed the revolutionary procedure to reach large concentration of specific DNA fragments, which is called Polymerase Chain Reaction or PCR. The PCR procedure allows scientists to copy and amplify specific regions of a DNA molecule (like genes) exponentially.
The first application of PCR was for analyzing the presence of genetic diseases mutations (genetic testing). A common application of PCR is the study of patterns of gene expression. Tissues and individual cells can be analyzed at different stages to see which genes are activated or inactivated, and quantitative PCR can be utilized to quantitate the actual levels of expression.Because PCR amplifies the regions of DNA that it targets, it can be used to analyze extremely small amounts of sample available. This is important for many different applications. For example, PCR may be used in phylogenetic analysis of ancient DNA such as that found in bones of human ancestors or frozen mammoth tissues. In forensic analysis, often there is only a trace amount of DNA available as evidence and PCR amplification solves this problem. Viral DNA can also be detected by PCR, but the primers used must be specific to the targeted sequences in the DNA of the virus. Furthermore, the high sensitivity of PCR permits virus detection soon after infection and sometimes even before disease onset. Such early detection may give physicians a significant lead time in treatment. PCR is also a critical tool to diagnose genetically rare diseases, such as Huntington disease.
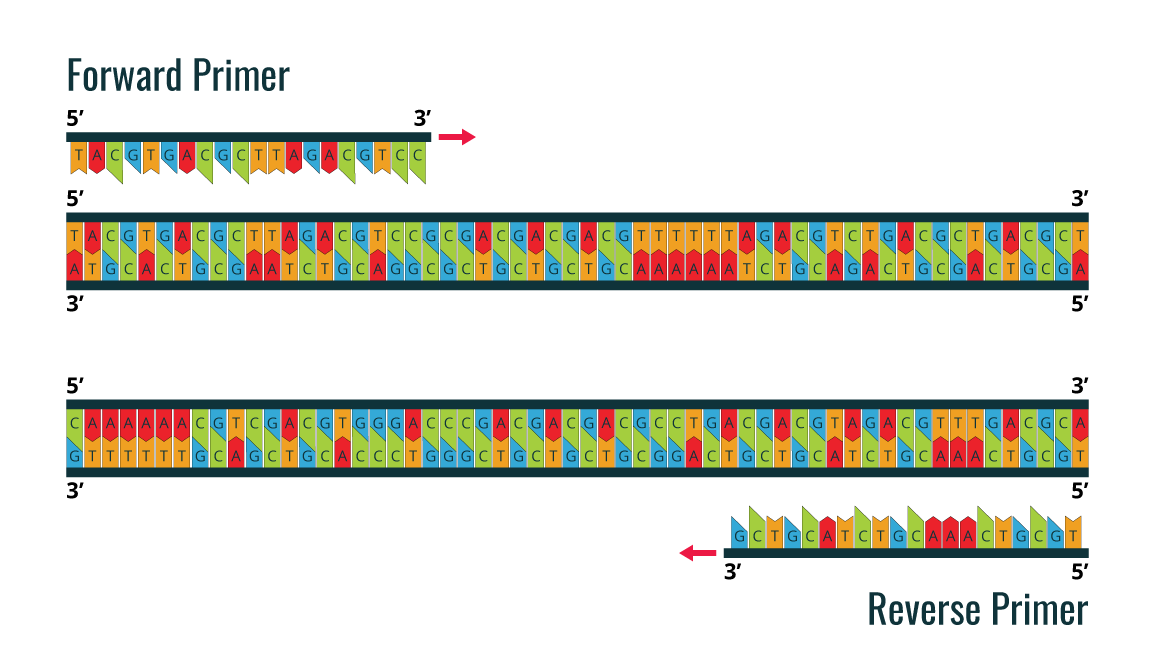
PCR utilizes the DNA polymerase enzyme, which naturally catalyzes the synthesis of DNA sequences. All DNA polymerases synthesize DNA in the 5’ to 3’ direction. In order to start the synthesis process, it is necessary to have a pair of chemically synthesized oligonucleotide primers made of DNA (as the primers are synthesized by cells). These two primers are designed to flank the DNA fragment, which will be amplified.
The image is an example of a forward primer and reverse primer, which are designed before PCR amplification is performed for the double stranded DNA sample template.
By using the ability of DNA polymerase to synthesize, PCR is a useful procedure in Molecular Biology. It requires four standard components and the 3-step processe: Denaturation, Annealing, and Extension

Upon high temperatures (around 95ºC) the DNA template molecule will denature and the two strands will move apart, breaking the hydrogen bonds that link the double helix DNA strands. Then, using lower temperatures, also called annealing temperatures (55ºC - 65ºC) will trigger primer annealing with the DNA template at the complementary sequences. At optimal temperature, extension occurs and DNA polymerase will then synthesize DNA, starting in the primer annealing region. After a certain period of time, dependent on DNA polymerase synthesis rate, the PCR solution is again heated at denaturing temperatures to repeat the previous cycle for several times.

What is amplification in PCR? It is a selective amplification of DNA or RNA targets using the polymerase chain reaction. Short single stranded primers are extended on the target template using repeated cycles of heat denaturation, annealing, and extension.
Since high temperatures are required to denature the dsDNA molecule, the DNA polymerase in solution has to be able to maintain its catalytic activity after such extreme temperatures. Thus, most of the DNA polymerases used inPCR protocols are extracted and purified from extremophilic microorganisms accustomed to living in high temperature environments. As examples, there are the Taq-polymerase from Thermophilus aquaticus or the Pfu-polymerase which is from Pyrococcus furiosus. These can handle temperatures of 95ºC and 100ºC, respectively. There are dozens of DNA polymerase used in PCR protocols, each one with its own specificity. Some of them have proofreading activity, others exonuclease activity, and more.
PCR is a relatively fast technique and a PCR cycle generally takes about 40 minutes to 1 hour to complete 40 cycles. At the end of each PCR cycle, the PCR product or amplicon will increase exponentially because the newly synthesized DNA sequences can be used as templates (in addition to the original DNA template). Usually, 20 to 30 standard PCR cycles are enough to promote an increment of 106 to 109 of the DNA fragments flanked by the two primers. The following image depicts how the PCR amplification process works to exponentially create copies of the DNA sample template.
Gel electrophoresis is one of the principal tools of molecular biology. The basic principle is that DNA, RNA, and proteins can all be separated by utilizing an electric field and their size. In gel electrophoresis, DNA and RNA can be separated on the basis of size, by running the genetic material through an electrically charged agarose gel. Larger sized molecules will travel through this gel at a different rate than smaller molecules and thus become physically and visually separated. Proteins can also be separated on the basis of size by using an SDS-PAGE gel, or on the basis of size and their electric charge by using 2D gel electrophoresis.
Depending on the expected size of the amplified fragment, a fraction of your PCR reaction should be loaded onto a 0.8–3% agarose gel containing 1 μg ml–1 ethidium bromide. Typically, one tenth or one-fifth of the reaction volume is loaded and the remainder is stored at 4°C or –20°C for future use. An aliquot of loading dye containing glycerol and a marker such as bromophenol blue should be added to the sample to assist both loading on the agarose gel and visualization of the sample migration through the gel. After gel electrophoresis is complete, the gel is visualized using UV light and special equipment. Non-toxic dyes such as SYBR® green can also be used to visualize the results. The intensity of the band can be used to estimate the amount of product of a particular molecular weight (relative to a molecular ladder). Gel electrophoresis also shows the specificity of the reaction, because the presence of multiple bands indicates secondary amplification products. A photo can be taken for this gel data, and then the results may be explained according to this data.
Go to Sample Preparation and Protocols section (PCR Product Analysis: Gel Electrophoresis) for more protocol details.Boster Bio provides a range of PCR products that includes DNA/RNA Extraction kits, PCR/RT-PCR kits, Master Mixes, etc. for your research needs.
Quantitative Reverse Transcription PCR is another approach in Molecular Biology resulted in highly specific targeted sequence. Comprises a set of procedures for quantification analysis includes Baseline correction, Threshold setting, Standard curve and Relative quantification. (Change the letter Q into small letter: qRT)
Advances in molecular biology allow us to diversify the applications for PCR procedure and the methods which molecular biologists study the genetic components of a cell. The reverse transcriptase enzyme is an enzyme that has a catalytic activity similar to DNA polymerase, but instead of DNA, it uses mRNA as the template to generate a DNA strand. This approach allows us to identify only genes that are being expressed in a specific cell at a specific time and environmental condition. After synthesis of complementary DNA or cDNA strand from the mRNA template by reverse transcriptase and using standard PCR protocols, it is possible to perform a qRT-PCR (quantitative real time PCR).
The qRT-PCR is based on the principle that higher or lower initial amounts of a specific DNA sequence will lead to higher or lower concentrations of amplicons respectively. Thus the PCR protocol will use a fluorescence dye to tag the newly synthesized sequences. Unlike a standard PCR, the qRT-PCR does not require purification and agarose gel analysis to analyze the amplicons. Since the amplicons are dyed, quantification of fluorescence will allow us to infer the amplicons concentrations. As an example, the SYBR green dye binds to dsDNA, but does not bind to single stranded DNA and RNA. When SYBR green dye is added to the PCR solution, it will radiate fluorescence only when it is bound to DNA, which uncovers the amplification of the DNA fragment flanked by the primers..
| Requirements | |
|---|---|
| Baseline Correction | In order to avoid variation in background signal caused by external factors not related to the samples (e.g. plasticware, light leaking, fluorescence probe not quenched, etc…), it is recommended to select the fluorescence intensity from the first cycles, usually cycles 5-15 and uncover a constant and linear component of background fluorescence. This information will then be used to define the baseline for analysis. |
| Threshold Setting | It is also important to select a quantification zone that is over the detection limit of the instrument used. Usually the number of cycles needed is directly proportional to the sample copy number. Instead of using the intensity of minimum fluorescence detected by the instrument, a quantification zone with higher fluorescence intensity is selected and a threshold defined for this area. |
| Standard Curve | The standard curve calibration is obtained based on the known concentration of the standards used. As recommended during qRT-PCR experiments, serial dilutions of a standard should be prepared. Then the Cq is determined for each dilution of the standard and plotted against the concentration. As a result, a standard curve will be displayed and used to infer about the concentration of the samples. The threshold should be the same for standard samples as well as for samples tested. |
| Relative Quantification | Usually relative quantification is used to make the data interpretation more intuitive and clear. Instead of plotting the Cq values for each sample, also denominated as absolute quantification, the Cq differences between samples are plotted, which allows us to access the expression fold changes for a specific targeted gene. |
You can access more information about sample preparation, protocols, and troubleshooting in our PCR Technical Resource Center.

PCR sample preparation is very crucial for reliable results. It includes accurate extraction and purification of DNA or RNA to the library preparation and screening.
Learn our PCR Sample Preparation
Learn stepwise PCR protocols from reagent preparation to data analysis. Check out their differences to learn how they compare and their advantages and disadvantage.

Get To Know Some PCR Troubleshooting Tips With This Guide. It Has Some Commonly Encountered Problems And Solutions To PCR.

Boster offers the best and reliable COVID-19 Detection Kits, Primers/probes, RNA Extraction Kits, and much more products at affordable prices. Get a quote today!