This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Epigenetic biomarker validation made easy
Epigenetic modifications are emerging as important diagnostic and prognostic biomarkers in many fields of medicine. Our Epigentics experts can help you stay ahead of the game. From genome-wide DNA methylation profiling to targeted pathway analysis, we can help you turn your samples into meaningful data.
Begin Inquiry
From sample and library preparation, sequencing and analysis, our epigenetics experts have
successfully helped our customers through these difficulties to save time, money and data.
We promise a concierge-like service every step of the way, turning your sample into quality
data to propel your Epigenetics research.
Our services include
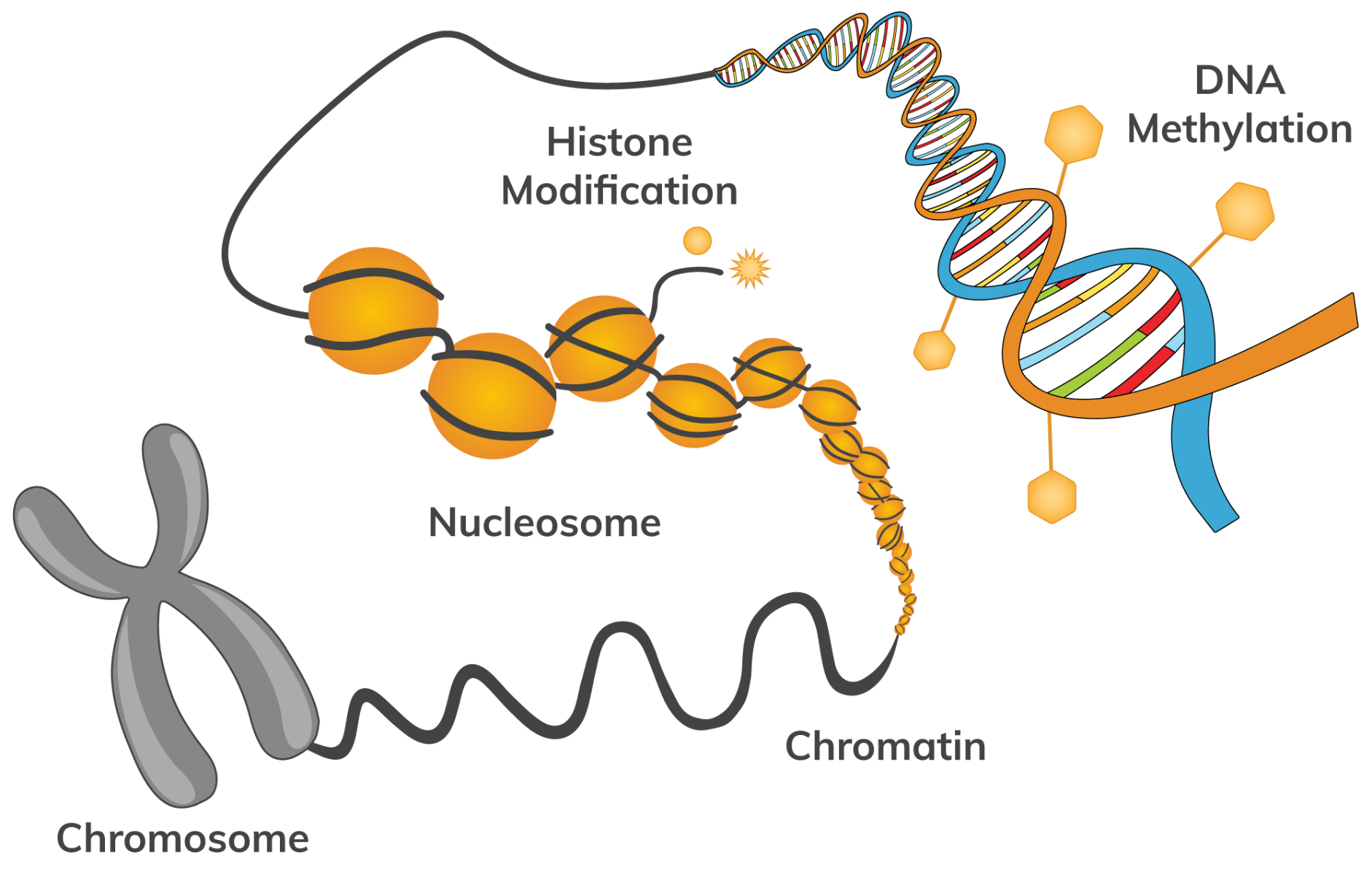
Epigenetics is the study of changes caused by the modification of gene expression which is caused by mechanisms other than in the underlying DNA. However, it is the epigenetic regulation of DNA that results in both phenotypic heritable traits and the production of distinguishing cell types, such as skin cells and liver cells. Epigenetics can change the way your body reads a DNA sequence via histone modifications or DNA methylation, but unlike genetic changes, it is possible for these to be reversed. Non-coding RNA expression can also regulate gene expression during or after transcription. By regulating gene expression in a coordinated fashion via patterns that can change over a lifetime, epigenetics can be termed as the study of how behavior and environment can affect the way our genes work.
Epigenetics can be used to understand genetic regulation, cellular differentiation, embryology, aging, and diseases such as cancer, and can also be used to explore novel avenues for targeted therapeutics or personalized medicines.
Genome-wide analysis and ChIP-seq techniques via NGS allow a better understanding of epigenetic events, to analyze DNA methylation, DNA demethylation, and the functional effects of these changes on cell development and differentiation, disease identification, and treatment.
As easy as 1, 2, 3.
DNA Methylation can’t be analyzed using standard sequencing methods because the methyl group is covalently bound to the cytosine (5-methylcytosine, 5-mC). So bisulfite sequencing has become the gold standard to make epigenetic biomarker validation simple. Bisulfite reagents convert unmethylated cytosine residues to uracils, leaving the methylated ones as is, so during PCR, the DNA polymerization recognizes the uracils as thymosine and the unmethylated cytosines as is. The final output is then compared to reference materials. In NGS methods, the bisulfite-converted DNA samples are constructed into libraries that contain adaptors for NGS sequencing. The most popular NGS methods include Reduced Representation Busilfute Sequencing (RRBS), Targeted Bisulfite Sequencing (TBS), and Whole Genome Bisulfite Sequencing (WGBS).
The main difference is the percentage of the total genome that is sequenced. If the customer is at discovery phase and has a limited budget, we usually recommend starting with RRBS or TBS since these 2 platforms enrich CpG dense regions. If you prefer to look at the entire genome or CHG and CHH context, WGBS would be a better option.
Genome coverage: RRBS < TBS < WGBS
Whether you need to validate methylation array data in a large sample cohort or investigate a specific gene region, our expert scientists can help design, validate and evaluate DNA methylation changes with targeted bisulfite sequencing.
Comprehensive and reliable DNA methylation sequencing analysis. Boster Bio’s DNA methylation services are genome-wide/whole-genome DNA methylation analysis at single nucleotide resolution, each designed to suit your specific coverage needs.
Methylated DNA Immunoprecipitation (MeDIP) is a bisulfite-free method for analyzing genome-wide methylation using antibodies targeting 5-mC to enrich for methylated DNA. This enriched fraction is then used in analysis or quantification via ELISA technique.
Services included in Basic analysis #1-5 and full analysis
Histone modifications to the core histone proteins can affect gene expression as well. This can be detected using Chromatin Immunoprecipitation (ChIP), where proteins are crosslinked to DNA and protein-specific antibodies are used to selectively precipitate bound DNA fragments to the protein. These enriched fragments can be sequenced via ChIP-seq NGS sequencing for a genome-wide profile of histone modification.
Boster Bio’s Chip-seq services cater for genome-wide mapping of histone modifications, protein DNA interactions, and identifying consensus protein binding sites in DNA for transcription factors or other enzymes.
The Assay for Transposase-Accessible Chromatin (ATAC-seq) is a derivative of ChIP-seq where the presence or absence of open chromatin is measured. This method uses transposase which can only insert adaptor sequences into open chromatin sites to distinguish unique binding sites or transcription factors bound within the native chromatin and to help us understand the role of chromatin structure changes.
Services Include
Accelerated biological aging can occur via the accumulation of epigenetic drift or mutations over time characterized by an increase in gene expression noise and can be associated with a number of diseases such as cancer and obesity-related illnesses. Biological age refers to epigenetic alternation and DNA methylation and can be determined by measuring DNA methylation at multiple sites.
Boster’s Epigenetic Aging Service utilizes the Simplified Whole-panel Amplification Reaction Method (SWARM®), which is a robust targeted bisulfite sequencing approach to deliver reproducible high-throughput methylation data. Applications include biomarker studies as well as arthritis, allergies, forensics, fertility, diabetes, alzheimers/parkinson’s and circadian rhythm research.
Services Include:
Mouse over one of the cards below to see more information

Boster Bio offers a full range of quality NGS services extending from nucleic acid extraction to bioinformatic analysis.
Boster Bio utilizes the latest tech and decades of experience to deliver high-quality gene expression profiling solutions.
Boster Bio’s experts have developed workflows for accurate microbiome quantification.
It is our project concierges' mission to make your project experience as smooth and memorable as possible. They are subject matter experts who are easily accessible around the clock, always happy to help you solve problems, make recommendations and sort through options.
End-to-end
and custom service
Optimised sample
processing workflows
Cutting-edge
bioinformatics
Competitive
pricing
Project concierge
to help at every step of the way
Fast sample
turnaround
End-to-end
and custom service
Optimised sample
processing workflows
Cutting-edge
bioinformatics
Competitive
pricing
Project concierge
to help at every step of the way
Fast sample
turnaround