This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Cellular senescence refers to a state where cells permanently cease to divide in response to stress or damage, while remaining metabolically active. It plays a crucial role in tumor suppression and tissue repair, but excessive accumulation of senescent cells can lead to tissue dysfunction and chronic inflammation.
Triggers of cellular senescence include telomere shortening, DNA damage, oxidative stress, and oncogene activation. These factors activate cell cycle checkpoints, leading to growth arrest. As organisms age, the accumulation of senescent cells is considered a significant contributor to aging and the development of age-related diseases such as atherosclerosis, diabetes, and neurodegenerative disorders.
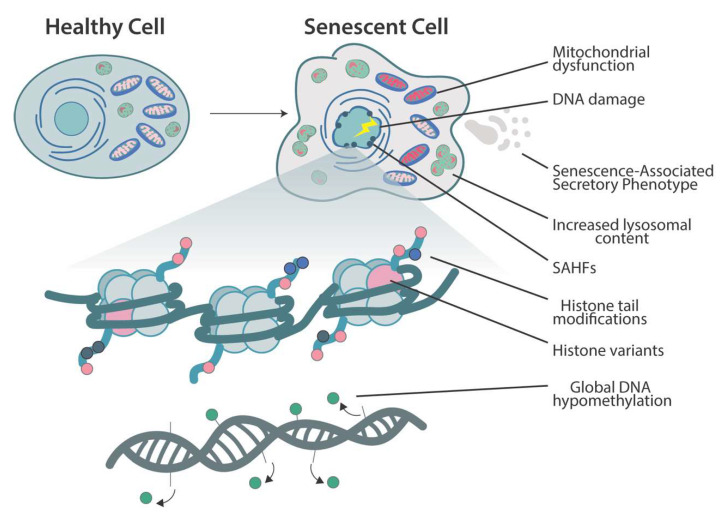
The process of cellular senescence involves:
Cellular senescence is a stress response that leads cells to enter a permanent state of growth arrest while maintaining metabolic activity. It represents a critical switch in cell fate that can be triggered by various intrinsic and extrinsic stressors. But why do cells undergo this transition, and what implications does it have for tissue homeostasis and disease?
Senescent cells are not merely passive bystanders; they play complex and context-dependent roles in both health and disease. Their functions can be categorized as beneficial, detrimental, or ambiguous depending on the biological setting.
During embryonic development, senescent cells contribute to tissue remodeling by secreting signaling molecules such as matrix metalloproteinases (MMP2, MMP9) and fibroblast growth factors (FGF4, FGF8). These factors coordinate cell migration and morphogenesis during organogenesis.
In wound healing and regeneration, senescent cells release platelet-derived growth factor AA (PDGF-AA) and other cytokines that recruit immune cells and fibroblasts to the injury site, promoting tissue repair.
Senescence also serves as a tumor-suppressive mechanism. The activation of p53, p21, and p16INK4a halts the proliferation of cells that exhibit DNA damage or oncogenic stress, thereby preventing malignant transformation.
Problems arise when senescent cells persist in tissues without being cleared. Chronically senescent cells secrete a complex mixture of inflammatory cytokines, chemokines, and proteases, collectively known as the senescence-associated secretory phenotype (SASP). Components such as interleukin-6 (IL-6), interleukin-1 receptor antagonist (IL-1RA), interferon-gamma (IFN-γ), growth-related oncogene alpha (GROα), and various MMPs contribute to a pro-tumorigenic microenvironment by promoting immune evasion and angiogenesis.
Senescent cells also contribute to stem cell exhaustion. Persistent expression of p16INK4a and p21 in the tissue microenvironment impairs the self-renewal capacity of stem cells, leading to reduced regenerative potential and accelerated aging.
Another key pathological role is in chronic inflammation. SASP factors such as IL-6 and IL-8 drive long-term immune activation, disrupting tissue architecture and function, and potentially contributing to age-related diseases.
Emerging research suggests that senescent cells might play a role in cellular reprogramming. Certain SASP components—particularly IL-6—have been implicated in facilitating dedifferentiation or transdifferentiation under specific conditions. However, the exact mechanisms remain unclear, and this process may be context-dependent: potentially beneficial for regeneration in one setting, yet harmful in another by promoting tumorigenesis.

Senescence is often initiated when cells accumulate persistent DNA damage, leading to genomic instability. Various stresses – critically short telomeres, oxidative or oncogenic stress, radiation, and chemical agents – generate DNA lesions that chronically activate the DNA damage response (DDR). Cells then halt the cell cycle to attempt repair, but if damage is irreparable, the DDR signal persists, enforcing a permanent arrest. In fact, recent reviews emphasize that “the principal cause of senescence is DNA damage,” which engages DDR kinases (ATM/ATR) and downstream effectors. Senescent cells are characterized by unresolved DNA double-strand breaks and a constitutive DDR: they exhibit repair foci (γH2AX, 53BP1) that do not resolve, high p53 activity and chromatin remodeling, all of which lock in the arrest.
In senescence, the DDR not only halts the cell cycle but also triggers the senescence program. DNA lesions activate checkpoint kinases (ATM/ATR → Chk1/Chk2), which induce cell-cycle inhibitors like p53 and p21. When damage is severe or chronic, these signals remain “on,” and the cell essentially treats its persistent damage as irreparable. This prolonged DDR is a hallmark of senescence: one review notes that senescent cells “undergo dramatic gene expression changes along with chromatin remodeling and engagement of a persistent DNA damage response”. In practice, stalled replication forks and telomeric breaks continually signal through DDR kinases, keeping p53–p21 engaged and enforcing arrest. Thus, a self-perpetuating DDR – often stemming from hard-to-repair lesions like double-strand breaks – is central to establishing senescence.

Telomeres, the protective caps of chromosomes, play a key role in senescence. With each cell division, telomeres shorten, and when they become critically short or uncapped, they resemble DNA double-strand breaks. This "end-replication problem" triggers a classic DDR that induces senescence.
As one recent review states:
"Telomere shortening and damage are recognized causes of cellular senescence and ageing."
In other words, telomere attrition chronically stimulates ATM/ATR signaling and the p53/p21 pathway, mirroring other forms of DNA damage. In aging tissues and replicative cultures, telomere-driven senescence is a major pathway to cell-cycle exit.
Thus, telomere dysfunction directly connects genomic instability to the senescence program.
After DNA damage, cells enforce checkpoint arrests to prevent proliferation. In senescence, these checkpoints become locked “on.” The canonical arrest occurs at G1: damaged cells stabilize p53, upregulate p21WAF1/CIP1, and inhibit cyclin/CDK complexes, preventing Rb phosphorylation and blocking the G1→S transition. This p53–p21 pathway is activated by various stresses (telomere attrition, oxidative or oncogenic signals) and, once chronically engaged, enforces an essentially irreversible arrest. In addition, the p16INK4a–Rb pathway provides a second barrier. Stress signals (e.g., aberrant mitogenic or oxidative cues) induce p16INK4a, which directly inhibits CDK4/6 and further activates Rb, reinforcing the G1 block. Together, these two tumor-suppressor circuits form a robust anti-proliferative barrier: senescent cells typically activate one or both pathways, and disabling p53 or p16 can delay or bypass the arrest.
Although senescence is most often a G1 arrest, evidence shows that prolonged G2/M checkpoint activation can also induce a senescent state. In cases of severe G2-specific DNA damage, cells cannot safely enter mitosis and instead exit the cell cycle from G2. For example, chronic DNA damage signaling keeps ATM/ATR and Chk1/Chk2 active, preventing activation of the mitotic Cdk1/cyclin B complex. Under these conditions, p21 can accumulate in G2 and inhibit mitotic CDKs, leading to a stable "G2 exit" arrest. In essence, the cell never completes mitosis and instead enters a tetraploid senescent-like state. This G2-based senescence is thought to occur in certain contexts (e.g., after intense genotoxic stress or in cancer therapy) and represents an alternative way to enforce a permanent arrest.
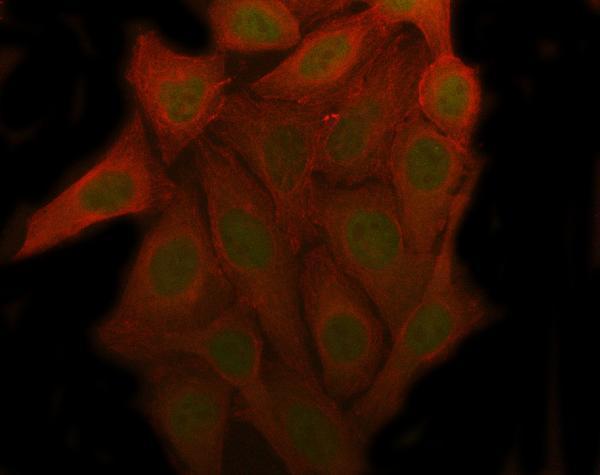
The p53–p21 and p16INK4a–Rb pathways are the final effectors of checkpoint control in senescence. DNA damage or telomere dysfunction activate p53, which transcribes p21. p21 inhibits cyclin E–Cdk2 and cyclin D–Cdk4/6, keeping Rb in its active (hypophosphorylated) state. In parallel, stress signals (including oncogenic signals) induce p16INK4a, which binds Cdk4/6 and also prevents Rb phosphorylation. The result is the same: active Rb suppresses E2F target genes required for S phase, locking the cell in G1. Thus, chronic activation of these pathways “triggers cellular senescence”. In practice, cells often use both circuits for added stability, and if one is lost (e.g. p53 mutation) the other can partially compensate. This dual checkpoint logic is why loss of p53 or p16 can enable cells to bypass senescence (at the cost of genomic stability).
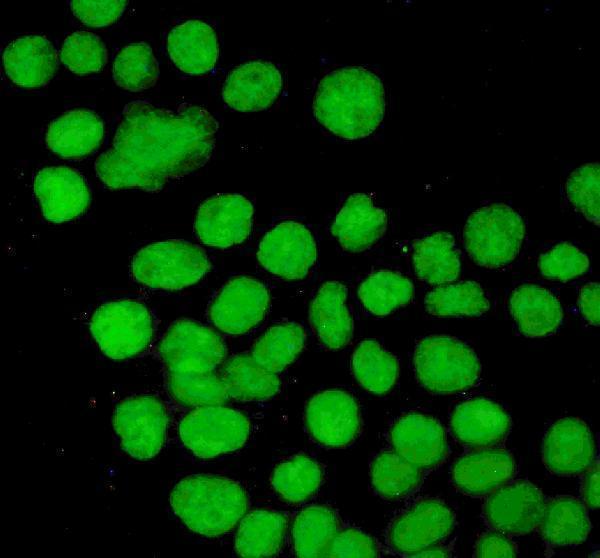
Senescence is accompanied by widespread epigenomic remodeling. The nuclear architecture changes dramatically: regions of heterochromatin compact into distinct Senescence-Associated Heterochromatin Foci (SAHF) that silence proliferation-promoting genes, while other parts of the genome lose heterochromatin. For example, large-scale chromatin reorganization occurs in senescence and global DNA hypomethylation is observed in replicative senescence. At the same time, specific histone marks are shuffled – for instance, loss of repressive H3K9me2/3 and gain of activating marks at SASP loci – and non-histone chromatin factors are redistributed.
In short, the chromatin landscape is reshaped: proliferation genes are epigenetically silenced, and many other genes (including stress response and secretory genes) become more accessible. This epigenetic reprogramming helps lock in the arrested state and define the senescent phenotype. Recent reviews emphasize that such epigenetic alterations are a hallmark of senescence: “senescent cell nuclei undergo epigenetic reprogramming” that regulates the SASP and the stability of arrest.
In practice, this means that senescence involves a transition to a distinct transcriptional state, supported by changes in DNA methylation, histone variants, and chromatin structure, which together enforce the non-proliferative program and its secretory output.
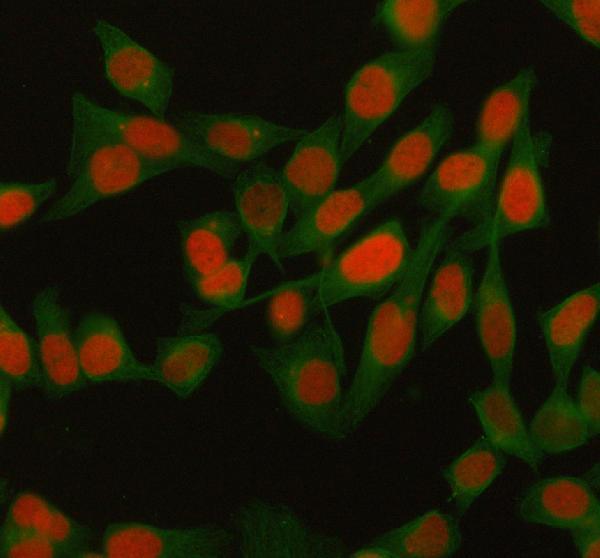
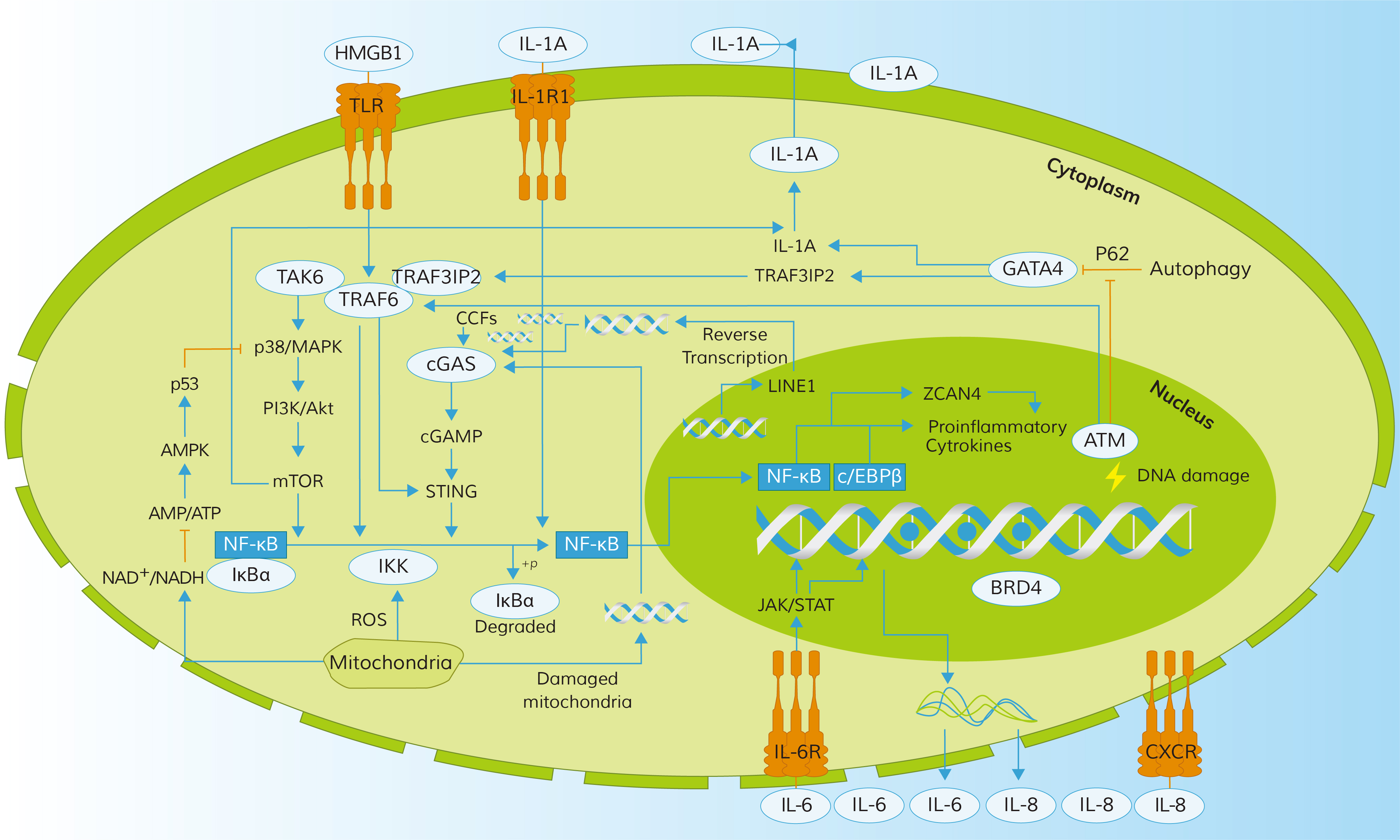
While cell division stops, senescent cells remain metabolically active and undergo striking functional changes. One hallmark is the Senescence-Associated Secretory Phenotype (SASP), a chronic inflammatory secretome. Senescent cells release a complex mix of cytokines (e.g. IL-6, IL-8), chemokines (e.g. CXCL1), growth factors (e.g. TGF-β, VEGF), and proteases (e.g. MMPs). This secretome can reinforce senescence through autocrine loops and signal neighboring cells to enter arrest or alter the tissue environment. Regulation of the SASP is tightly linked to the persistent DDR and specific signaling pathways: for example, unrepaired DNA damage leads to the formation of cytoplasmic chromatin fragments that activate cGAS–STING, in turn inducing NF-κB and C/EBPβ to drive SASP gene expression. The net effect is a robust inflammatory response: senescent fibroblasts, for instance, secrete IL-6 and IL-8 that can promote neighboring cell proliferation or tumor progression. The SASP thus has both beneficial and deleterious roles – it aids in wound healing and immune clearance of damaged cells but also contributes to chronic inflammation (termed “inflammaging”) and age-related pathology when senescent cells accumulate.

Senescent cells also exhibit characteristic morphological and metabolic changes. They enlarge and flatten dramatically, often becoming multinucleated, with increased cytoplasmic vacuoles and lysosomes (underlying the classic SA-β-galactosidase staining).
At the organelle level, mitochondria show marked dysfunction: their biogenesis and mitophagy are impaired, leading to the accumulation of damaged mitochondria that produce excess reactive oxygen species and have lower membrane potential.
In parallel, the endoplasmic reticulum and Golgi may expand to handle the secretory load, and lysosomal content increases.
These shifts reflect a broad metabolic reprogramming – senescent cells rely more on glycolysis and other pathways to fuel the SASP and stress responses.
In summary, the arrested state is coupled to a hypertrophic, pro-inflammatory phenotype: enlarged cell size, altered organelle function, and metabolic stress, all of which distinguish senescent cells from quiescent or dividing cells.

Cellular senescence is a complex biological process characterized by a stable cell cycle arrest in response to various stressors, including DNA damage, oxidative stress, and oncogenic signals. While senescence serves as a critical tumor suppressive mechanism, its accumulation over time contributes to aging and the pathogenesis of age-related diseases.
The accumulation of senescent cells in tissues is a hallmark of aging. These cells exhibit the senescence-associated secretory phenotype (SASP), releasing pro-inflammatory cytokines, chemokines, growth factors, and proteases that can disrupt tissue structure and function. For instance, in the cardiovascular system, senescent endothelial cells contribute to vascular stiffness and atherosclerosis. In the musculoskeletal system, senescent cells in joints are implicated in osteoarthritis development. Moreover, the presence of senescent cells in adipose tissue is associated with metabolic dysregulation and insulin resistance.
Beyond aging, senescence plays a dual role in disease. On the one hand, it acts as a barrier to tumorigenesis by halting the proliferation of damaged cells. On the other hand, the chronic presence of senescent cells and their SASP can promote a pro-inflammatory environment conducive to disease progression.
In cancer, while senescence prevents the growth of potential tumor cells, the SASP can paradoxically support tumor development by promoting angiogenesis, epithelial-to-mesenchymal transition, and immune evasion. In the context of cardiovascular diseases, senescent cells contribute to myocardial remodeling and heart failure. Moreover, the SASP has been implicated in the progression of neurodegenerative diseases, such as Alzheimer's disease, by exacerbating neuroinflammation.
Therapeutic strategies targeting senescent cells, known as senolytics, aim to eliminate these dysfunctional cells to alleviate their deleterious effects. Compounds like dasatinib and quercetin have shown promise in reducing senescent cell burden and improving tissue function in preclinical models.
Cellular senescence is no longer viewed as a mere byproduct of aging—it's now recognized as a key player in development, tissue repair, cancer biology, and age-related disease. As our understanding deepens, so does the need for high-quality reagents to study this complex process.
Whether you're investigating DNA damage response, checkpoint pathways, chromatin remodeling, or the SASP, reliable antibodies are critical for detecting key proteins like p53, p21, p16, γH2AX, IL-6, and more. At Boster Bio, we understand the technical demands of senescence research. That’s why we offer a growing collection of validated antibodies and ELISA kits designed to support cutting-edge studies in aging, oncology, and regenerative biology.
If you’re working on senescence, don’t let unreliable reagents slow your progress. Browse our catalog and discover how Boster’s rigorously tested products can help you generate publication-ready results with confidence.