This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Dilution ratio describes a simple dilution – a unit volume of solute (or sample) is combined with a desired unit volume of solvent (or diluent), to reach a desired total volume (Vsolute + Vsolvent = Total Vsolution)
Thus, a dilution ratio of 1:4 describes 1 part solute + 4 parts solvent = 5 parts total. The sum of both solute plus solvent equals total, f...


High background in DAB staining usually shows up as one of three patterns: a global brown/gray haze, edge-darkening, or granular brown speckling. The fastest way to fix it is to stop guessing and first identify which layer is generating the background: tissue chemistry, primary binding, the HRP detection/amplification layer, or...

ELISA results can look “clean”—tight duplicates and a smooth standard curve—and still be misleading. In practice, the most common failure is not pipetting technique, but controls that do not isolate the specific failure mode (system background, non-specific binding, matrix interference, or out-of-range samples). If you want a broader setup framework before drilling into controls, Boster’s ELISA experimental design checklist is a useful companion read.
This post focuses on four controls that most reliably de-risk interpretation:

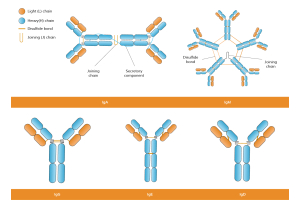
Microbial expression systems use engineered microbial organisms, such as bacteria or yeast, for the expression of antibody fragments quickly, efficiently, and at scale. By stripping away the complexities of mammalian cell culture, these systems can deliver functional proteins in days instead of weeks, making them a valuable complement to custom antibody production in mammalian systems. Formats like the Fab fragment, F(ab’)₂, scFv fragments, and VHH single-chain antibodies are particularly well-suited to microbial production because of their small size, simpler protein folding requirements, and lack of dependence on mammalian N-linked glycosylation. When Fc domain functions or full glycan structures are unnecessary, bacterial production in hosts like Escherichia coli or yeast secretory expression in Pichia pastoris offers a streamlined path from gene expression to purified reagent.
Antibody engineering has moved far beyond hybridoma technology and transgenic animal production. Today, recombinant protein production in microbial hosts provides a flexible and accessible option for generating high-quality antibody fragments. These truncated antibody molecules retain their antigen binding sites while omitting constant domains such as the Fc domain, reducing molecular weight and improving tissue penetration. This makes them highly valuable for diagnostics, targeted therapeutic proteins, and research tools.
Unlike mammalian cells or insect cell lines using the Baculovirus expression system, microbes like Escherichia coli grow rapidly, require inexpensive cell culture media, and can be genetically modified with precise recombinant DNA methods. For labs under tight deadlines, gene expression using Escherichia coli cells can turn a DNA sequence into a purified recombinant antibody fragment in a short timeframe. For those who need secretion and partial posttranslational changes, Pichia pastoris and Saccharomyces cerevisiae provide robust expression systems with secretion into the secretory pathway, simplifying recovery.
Choosing the right host involves balancing yield, protein expression quality, and downstream requirements. Each host type has specific advantages and limitations.
Escherichia coli is the most widely used Gram-negative bacteria for recombinant protein expression, particularly for scFv fragments, single-chain fragment variable formats, and Fab fragments. Its periplasmic expression pathway supports disulfide bonds crucial for antigen binding stability. For challenging constructs, molecular chaperones such as protein disulfide isomerase and other chaperone proteins can improve folding efficiency and reduce protein aggregation. While E. coli has limited glycosylation capabilities compared to mammalian, most antibody Fab fragments and fusion proteins do not require glycan modifications, making it ideal for bacterial production of non-glycosylated therapeutic proteins.
Pichia pastoris and Saccharomyces cerevisiae are leading yeast expression systems for antibody fragments that benefit from secretion. Pichia pastoris offers high-density fermentation and yeast secretory expression into the culture medium, easing purification. It can add simple glycans via its glycosylation machinery, though these differ from mammalian glycosylation sites. Glycoengineered Pichia pastoris strains can be used when partial glycosylation pathways are desirable. Saccharomyces cerevisiae is less common for high-titer production but offers well-developed strain engineering tools and synthetic inducible promoters for controlled gene expression.
Specialty systems like Bacillus subtilis can secrete proteins directly into the medium and are naturally free of endotoxins, making them attractive for sensitive diagnostic applications. Filamentous fungi are valued for their ability to support complex protein folding and post-translational modifications, but in recombinant protein production they remain less widely adopted than hosts like E. coli and Pichia pastoris, partly due to slower growth and historically fewer advanced genetic engineering tools—ongoing developments are reducing these limitations.
Despite differences in host biology, microbial production follows a predictable sequence.
The workflow begins with obtaining DNA constructs encoding the target fragment, sourced from hybridoma technology, phage display libraries, or synthetic gene design. These sequences are inserted into expression vectors tailored to the host organism’s codon usage and optimized to avoid unfavorable mRNA secondary structures. Vectors typically include strong promoters, secretion signal peptides for yeast-based systems, and affinity tags to facilitate purification. For Escherichia coli, the T7 promoter is widely used, whereas in Pichia pastoris, the AOX1 promoter enables methanol-inducible expression.
Once the vector is ready, it is introduced into the host cells—commonly via chemical transformation or electroporation in E. coli and electroporation in P. pastoris. Gene expression is then induced under host-specific conditions: isopropyl β-D-1-thiogalactopyranoside (IPTG) induction in E. coli or methanol feeding in Pichia. Throughout this stage, fermentation parameters such as temperature, pH, and dissolved oxygen are closely monitored to support optimal protein yield.
Proper folding is essential for preserving antigen-binding activity. In E. coli, directing expression to the periplasm facilitates disulfide bond formation, while co-expression with molecular chaperones can reduce aggregation. In yeast systems, secretion through the endoplasmic reticulum exposes the protein to endogenous chaperones, promoting correct folding and post-translational modifications.
Scaling up production requires fine-tuning fermentation strategies. In E. coli, fed-batch fermentation maintains high cell densities and consistent productivity. In P. pastoris, managing the methanol utilization pathway is critical, as excessive methanol can cause stress and reduce yield. Across both systems, minimizing cellular stress is key to sustaining performance.
Downstream processing often begins with affinity chromatography—commonly using His-tags, Protein L, or antigen-specific ligands—to isolate the antibody fragment from the culture medium. This is followed by polishing steps such as ion-exchange or size-exclusion chromatography to remove host cell contaminants. For bacterial systems, additional endotoxin removal steps are required to meet therapeutic-grade standards.
The final stage verifies that the product meets functional and regulatory requirements. Analytical assessments include SDS-PAGE for purity, mass spectrometry for identity, and binding assays such as ELISA or Western blot for functional performance. Stability testing ensures that the antibody fragment maintains its activity over time, supporting consistent therapeutic or diagnostic application.
Microbial expression systems enable the production of antibody fragments for a range of uses, spanning diagnostics, therapeutics, and research.
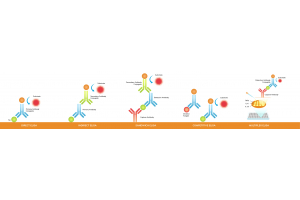
In assays such as ELISA, lateral flow devices, and biosensors, smaller antibody fragments can be densely immobilized on detection surfaces, enhancing signal sensitivity. Microbial production offers a rapid, cost-effective supply for these applications.
In the early days of custom antibody production, scientists explored various animal cell culture technology platforms to identify the ideal system
Growing demand for high-quality antibodies in therapeutics, diagnostics, and research is pushing innovation beyond traditional hybridoma technology and phage display methods. Single B cell antibody discovery addresses this need by enabling the direct isolation of antigen-specific B cells from immunized or naturally exposed subjects while preserving the native pairing of heavy and light chains, an approach that complements custom antibody production. This capability allows for the rapid generation of functional antibodies with greater diversity, including those targeting molecules that are difficult to address using conventional techniques.
Single B cell antibody discovery is a high-resolution single B cell screening technique that identifies and clones antibody sequences... from individual B cells. By isolating single antibody-secreting B cells and sequencing
Multiplex immunofluorescence (multiplex IF) rarely fails because you “missed a step.” It fails because you can’t reliably separate bleed-through, background, and true signal—and you don’t have a fast way to prove what’s actually happening.
Most multiplex problems are solved faster when you diagnose first: run the minimum controls, apply a few quick checks, then change the right lever in the right order. If you’re new to IF terminology, start here: immunofluorescence glossary. If you need a step-by-step workflow reference, use this resource (this post won’t repeat it): IHC/ICC/IF protocol resource.
To troubleshoot multiplex IF quickly: (1) run single-stain controls and view each single stain across all channels to detect bleed-through, (2) run a no-primary control to identify system/sample background, (3) check for saturation in any channel, and (4) optimize each channel for best signal-to-background—but keep settings consistent within the same channel when comparing conditions. Then adjust only one staining variable at a time.
Most multiplex issues fall into one (or more) of these patterns:
When you see these, don’t change everything. Start by proving what kind of problem it is.
You don’t need a dozen controls. You need the right ones.
What it is: Stain each target one at a time, using the same imaging settings you plan to use for multiplex.
What it tells you:
Use it like this (quick):
What it is: Run the full workflow without primary antibodies.
What it tells you:
What it is: Image only the brightest marker (or brightest single-stain) at the exposures you’re using for multiplex.
What it tells you:
Optional: Isotype control—useful when you suspect non-specific binding and your no-primary control is clean but staining still looks wrong. Don’t default to it as a first-line control.
| Quick check | When you’ll see it | Do this (fast) | What it means | Fix first |
|---|---|---|---|---|
| A) Saturation check | “Co-localization everywhere”, flat/glowy signal | Look for clipped highlights (max intensity) in any channel | Saturation can create false positives and fake overlap | Lower exposure/gain on the saturated channel before anything else |
| B) Cross-channel leak check (single-stain scan) | Signal shows up in multiple channels | View a single-stain image across all channels using multiplex settings | Spillover/bleed-through or detection cross-talk (not biology) | Reduce bright-channel exposure and rebalance signal; verify again with single-stain |
| C) Background source check (no-primary) | Haze/grain, especially in one channel | Compare no-primary to multiplex using the same display scaling | Background is system/sample/detection-driven | Tighten wash consistency; reduce non-specific signal sources; check detection/secondary behavior |
| D) Exposure consistency check | Overlap appears/disappears when brightness changes | Set each channel independently for clean signal-to-background, but keep the same settings within each channel when comparing conditions; avoid “auto” adjustments. | Imaging settings are driving the interpretation | Keep per-channel settings consistent for comparisons; re-evaluate overlap after settings are standardized. |
Rule of thumb: Fix imaging QC (saturation + per-channel exposure rules) before changing staining variables.
When multiplex looks wrong, apply fixes in this order (least effort → biggest impact):
Further reading (no overlap with this post): How to Choose Fluorophores for Multiplex IF. If what you’re seeing looks like tissue autofluorescence (broad, structure-like background), use this guide: 5 Tips to Reduce Autofluorescence.
| What |
|---|

Antibody phage display is a molecular screening technique used to identify high-affinity antibodies by displaying antibody fragments on the surface of bacteriophages. This method bypasses the need for animal immunization and allows for controlled, high-throughput screening of antibody–antigen interactions. Its versatility makes it a core tool in therapeutic antibody discovery, diagnostics, and research reagent development.
Phage display libraries are collections of bacteriophages engineered to present a vast diversity of antibody fragments, commonly single-chain variable fragments (scFv) or Fab fragments on their surfaces. These libraries contain millions to billions of unique variants.
Understanding the source and library design of a phage display library is critical, as it influences the diversity, specificity, and affinity of the resulting antibodies, a process closely aligned with custom antibody production strategies.
Phage libraries are typically constructed using:
These libraries serve as a starting point for selecting antibody candidates that specifically bind a target antigen.
Phage display screening follows a systematic cycle known as biopanning to isolate antigen-specific binders.
This section walks through the biopanning steps used to identify functional antibody fragments from a library.
Phage display enables fine control over screening conditions, allowing optimization for specificity, affinity, and even conformational epitope targeting.
Phage display offers powerful capabilities for antibody discovery, but it also comes with technical and biological considerations that affect its performance and suitability for specific applications.
Phage display offers several practical and technical benefits, particularly for researchers seeking high-throughput screening and control over antibody selection.
One of the main strengths of phage display is the ability to control the entire screening environment. Researchers can adjust pH, salt concentration, temperature, or antigen presentation format to select antibodies with specific properties, such as pH-resistance, epitope specificity, or stability under assay conditions. This level of precision is difficult to achieve using animal-based immunization methods.
Phage libraries can be constructed with sizes ranging from 107 to more than 1010 unique variants. This large diversity increases the chance of identifying rare antibody clones that bind novel, weakly immunogenic, or conformationally restricted epitopes. The diversity can be sourced from natural repertoires or synthetically designed CDR regions using amino acids tailored for function.
Because the entire process takes place in vitro, phage display eliminates the need for animal immunization. This makes it especially advantageous for generating antibodies against antigens that are toxic, non-immunogenic, or highly conserved across species. It also allows for rapid and ethical screening in regulatory-sensitive projects.
Phage display workflows are highly adaptable to robotic automation and scalable to industrial levels. From library construction to panning, expression, and screening, each step can be standardized and integrated into high-throughput systems. This makes it well suited for both academic discovery pipelines and commercial therapeutic development.
Unlike antibodies derived from immunized animals or B-cell based discovery platforms, phage display antibodies do not undergo somatic hypermutation or selection within a living organism. As a result, initial clones may lack the high affinity and specificity seen in naturally matured antibodies. Affinity maturation techniques such as error-prone PCR or chain shuffling are often needed to enhance binding strength.
The antibodies retrieved from naïve or synthetic libraries frequently show modest binding affinities at the screening stage. While functional, they may require additional optimization through directed evolution or site-directed mutagenesis to meet therapeutic or diagnostic thresholds.
Phage display is one of several antibody discovery methods, each with specific use cases.
This section contrasts phage display with hybridoma, single B cell, and Plasma Cell Discovery (PCD) technologies.
| Method | Screening Scale | Affinity Potential | Time to Results | Best Suited For |
|---|---|---|---|---|
| Phage Display | 109–1010 clones | Moderate | Moderate | Broad antigen coverage |
| Hybridoma | 103–104 clones | High (in vivo) | Slow | Research reagents |
| Single B Cell | 104–105 clones | High | Fast | Therapeutic antibody development |
| PCD (Boster Bio) | Up to whole spleen | Very High | Moderate | Diagnostics and difficult targets |
While phage display offers broader library sizes, technologies like single B cell and Plasma Cell Discovery (PCD) benefit from natural affinity maturation.
Phage display has proven to be a valuable tool across research, diagnostic, and therapeutic fields. Its flexibility and ability to generate high-specificity antibody fragments make it well-suited for various applications throughout the drug discovery and development pipeline.
Several clinically approved monoclonal antibodies originated from phage display screening, including adalimumab (Humira), a fully human anti-TNFα antibody used to treat autoimmune diseases such as rheumatoid arthritis and Crohn's disease. Phage display allows for the early selection of high-affinity clones that can be further engineered, humanized, and optimized for clinical use.
Phage-derived antibodies are commonly used in diagnostic platforms such as ELISA kits, lateral flow assays (LFAs), and biosensors. These antibodies offer high batch-to-batch consistency, essential for reliable test performance in clinical settings. Their specificity makes them suitable for detecting biomarkers in complex biological samples.
In academic and preclinical research, phage display–derived antibodies play a central role in target validation, protein–protein interaction studies, receptor mapping, and in vivo imaging. Researchers can rapidly isolate antibody fragments with desired binding characteristics, making phage display a practical choice for screening and functional studies.
Phage display fits naturally into modern biologics development workflows. Because antibody fragments can be recovered as DNA sequences, they are easily modified using molecular biology techniques. These include affinity maturation, isotype switching, Fc engineering, and conversion into full-length IgGs or bispecific formats.
Optimizing phage display screening involves more than just running standard protocols. Each step, from antigen preparation to binder validation, must be carefully designed to improve the chances of identifying functional, application-ready antibodies.
Antigen quality directly influences the quality of binders retrieved from the phage display library. Whenever possible, antigens should be presented in their native conformation, particularly when conformational epitopes are important. For proteins, consider using eukaryotic expression systems to preserve post-translational modifications. Immobilization methods should also minimize structural distortion, and antigen orientation on the capture surface should be consistent and accessible.
To reduce the likelihood of isolating non-specific binders, incorporate subtractive panning or negative selection. This involves exposing the phage library to irrelevant proteins, carrier molecules, or closely related antigens before positive selection. This step helps remove clones that bind to common protein scaffolds or matrix components, thereby enriching for clones with true target specificity.
After several rounds of panning, it is important to analyze the pool of enriched clones. Sequencing a diverse set of phage clones helps identify whether certain sequences are being preferentially selected, which can indicate convergence toward high-affinity binders. This insight can guide clone prioritization for downstream validation and highlight structural motifs worth engineering.
Once promising binders are identified, they should be reformatted from their original scFv or Fab fragment into full-length IgG or other desired formats. This step is crucial because binding behavior can change upon reformatting. Validate these reformatted antibodies across key assays such as ELISA, Western blotting, and immunohistochemistry to confirm performance and specificity under real-world conditions.
Phage display remains a foundational technology for antibody generation, particularly when animal immunization is not viable or when in vitro control is preferred. Although newer methods like Plasma Cell Discovery (PCD) show promise with higher hit rates and in vivo maturation, phage display remains indispensable for early-stage research and rapid screening campaigns.
Need help planning your antibody discovery project? Explore how Boster Bio can support your antibody discovery project with phage display, hybridoma development, and plasma cell-based screening. Our team provides recombinant antibody expression and validation for IHC, ELISA, WB, and therapeutic workflows.
Contact us today to discuss your project.