This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Multiplex immunofluorescence (multiplex IF) rarely fails because you “missed a step.” It fails because you can’t reliably separate bleed-through, background, and true signal—and you don’t have a fast way to prove what’s actually happening.
Most multiplex problems are solved faster when you diagnose first: run the minimum controls, apply a few quick checks, then change the right lever in the right order. If you’re new to IF terminology, start here: immunofluorescence glossary. If you need a step-by-step workflow reference, use this resource (this post won’t repeat it): IHC/ICC/IF protocol resource.
To troubleshoot multiplex IF quickly: (1) run single-stain controls and view each single stain across all channels to detect bleed-through, (2) run a no-primary control to identify system/sample background, (3) check for saturation in any channel, and (4) optimize each channel for best signal-to-background—but keep settings consistent within the same channel when comparing conditions. Then adjust only one staining variable at a time.
Most multiplex issues fall into one (or more) of these patterns:
When you see these, don’t change everything. Start by proving what kind of problem it is.
You don’t need a dozen controls. You need the right ones.
What it is: Stain each target one at a time, using the same imaging settings you plan to use for multiplex.
What it tells you:
Use it like this (quick):
What it is: Run the full workflow without primary antibodies.
What it tells you:
What it is: Image only the brightest marker (or brightest single-stain) at the exposures you’re using for multiplex.
What it tells you:
Optional: Isotype control—useful when you suspect non-specific binding and your no-primary control is clean but staining still looks wrong. Don’t default to it as a first-line control.
| Quick check | When you’ll see it | Do this (fast) | What it means | Fix first |
|---|---|---|---|---|
| A) Saturation check | “Co-localization everywhere”, flat/glowy signal | Look for clipped highlights (max intensity) in any channel | Saturation can create false positives and fake overlap | Lower exposure/gain on the saturated channel before anything else |
| B) Cross-channel leak check (single-stain scan) | Signal shows up in multiple channels | View a single-stain image across all channels using multiplex settings | Spillover/bleed-through or detection cross-talk (not biology) | Reduce bright-channel exposure and rebalance signal; verify again with single-stain |
| C) Background source check (no-primary) | Haze/grain, especially in one channel | Compare no-primary to multiplex using the same display scaling | Background is system/sample/detection-driven | Tighten wash consistency; reduce non-specific signal sources; check detection/secondary behavior |
| D) Exposure consistency check | Overlap appears/disappears when brightness changes | Set each channel independently for clean signal-to-background, but keep the same settings within each channel when comparing conditions; avoid “auto” adjustments. | Imaging settings are driving the interpretation | Keep per-channel settings consistent for comparisons; re-evaluate overlap after settings are standardized. |
Rule of thumb: Fix imaging QC (saturation + per-channel exposure rules) before changing staining variables.
When multiplex looks wrong, apply fixes in this order (least effort → biggest impact):
Further reading (no overlap with this post): How to Choose Fluorophores for Multiplex IF. If what you’re seeing looks like tissue autofluorescence (broad, structure-like background), use this guide: 5 Tips to Reduce Autofluorescence.
| What |
|---|

Antibody phage display is a molecular screening technique used to identify high-affinity antibodies by displaying antibody fragments on the surface of bacteriophages. This method bypasses the need for animal immunization and allows for controlled, high-throughput screening of antibody–antigen interactions. Its versatility makes it a core tool in therapeutic antibody discovery, diagnostics, and research reagent development.
Phage display libraries are collections of bacteriophages engineered to present a vast diversity of antibody fragments, commonly single-chain variable fragments (scFv) or Fab fragments on their surfaces. These libraries contain millions to billions of unique variants.
Understanding the source and library design of a phage display library is critical, as it influences the diversity, specificity, and affinity of the resulting antibodies, a process closely aligned with custom antibody production strategies.
Phage libraries are typically constructed using:
These libraries serve as a starting point for selecting antibody candidates that specifically bind a target antigen.
Phage display screening follows a systematic cycle known as biopanning to isolate antigen-specific binders.
This section walks through the biopanning steps used to identify functional antibody fragments from a library.
Phage display enables fine control over screening conditions, allowing optimization for specificity, affinity, and even conformational epitope targeting.
Phage display offers powerful capabilities for antibody discovery, but it also comes with technical and biological considerations that affect its performance and suitability for specific applications.
Phage display offers several practical and technical benefits, particularly for researchers seeking high-throughput screening and control over antibody selection.
One of the main strengths of phage display is the ability to control the entire screening environment. Researchers can adjust pH, salt concentration, temperature, or antigen presentation format to select antibodies with specific properties, such as pH-resistance, epitope specificity, or stability under assay conditions. This level of precision is difficult to achieve using animal-based immunization methods.
Phage libraries can be constructed with sizes ranging from 107 to more than 1010 unique variants. This large diversity increases the chance of identifying rare antibody clones that bind novel, weakly immunogenic, or conformationally restricted epitopes. The diversity can be sourced from natural repertoires or synthetically designed CDR regions using amino acids tailored for function.
Because the entire process takes place in vitro, phage display eliminates the need for animal immunization. This makes it especially advantageous for generating antibodies against antigens that are toxic, non-immunogenic, or highly conserved across species. It also allows for rapid and ethical screening in regulatory-sensitive projects.
Phage display workflows are highly adaptable to robotic automation and scalable to industrial levels. From library construction to panning, expression, and screening, each step can be standardized and integrated into high-throughput systems. This makes it well suited for both academic discovery pipelines and commercial therapeutic development.
Unlike antibodies derived from immunized animals or B-cell based discovery platforms, phage display antibodies do not undergo somatic hypermutation or selection within a living organism. As a result, initial clones may lack the high affinity and specificity seen in naturally matured antibodies. Affinity maturation techniques such as error-prone PCR or chain shuffling are often needed to enhance binding strength.
The antibodies retrieved from naïve or synthetic libraries frequently show modest binding affinities at the screening stage. While functional, they may require additional optimization through directed evolution or site-directed mutagenesis to meet therapeutic or diagnostic thresholds.
Phage display is one of several antibody discovery methods, each with specific use cases.
This section contrasts phage display with hybridoma, single B cell, and Plasma Cell Discovery (PCD) technologies.
| Method | Screening Scale | Affinity Potential | Time to Results | Best Suited For |
|---|---|---|---|---|
| Phage Display | 109–1010 clones | Moderate | Moderate | Broad antigen coverage |
| Hybridoma | 103–104 clones | High (in vivo) | Slow | Research reagents |
| Single B Cell | 104–105 clones | High | Fast | Therapeutic antibody development |
| PCD (Boster Bio) | Up to whole spleen | Very High | Moderate | Diagnostics and difficult targets |
While phage display offers broader library sizes, technologies like single B cell and Plasma Cell Discovery (PCD) benefit from natural affinity maturation.
Phage display has proven to be a valuable tool across research, diagnostic, and therapeutic fields. Its flexibility and ability to generate high-specificity antibody fragments make it well-suited for various applications throughout the drug discovery and development pipeline.
Several clinically approved monoclonal antibodies originated from phage display screening, including adalimumab (Humira), a fully human anti-TNFα antibody used to treat autoimmune diseases such as rheumatoid arthritis and Crohn's disease. Phage display allows for the early selection of high-affinity clones that can be further engineered, humanized, and optimized for clinical use.
Phage-derived antibodies are commonly used in diagnostic platforms such as ELISA kits, lateral flow assays (LFAs), and biosensors. These antibodies offer high batch-to-batch consistency, essential for reliable test performance in clinical settings. Their specificity makes them suitable for detecting biomarkers in complex biological samples.
In academic and preclinical research, phage display–derived antibodies play a central role in target validation, protein–protein interaction studies, receptor mapping, and in vivo imaging. Researchers can rapidly isolate antibody fragments with desired binding characteristics, making phage display a practical choice for screening and functional studies.
Phage display fits naturally into modern biologics development workflows. Because antibody fragments can be recovered as DNA sequences, they are easily modified using molecular biology techniques. These include affinity maturation, isotype switching, Fc engineering, and conversion into full-length IgGs or bispecific formats.
Optimizing phage display screening involves more than just running standard protocols. Each step, from antigen preparation to binder validation, must be carefully designed to improve the chances of identifying functional, application-ready antibodies.
Antigen quality directly influences the quality of binders retrieved from the phage display library. Whenever possible, antigens should be presented in their native conformation, particularly when conformational epitopes are important. For proteins, consider using eukaryotic expression systems to preserve post-translational modifications. Immobilization methods should also minimize structural distortion, and antigen orientation on the capture surface should be consistent and accessible.
To reduce the likelihood of isolating non-specific binders, incorporate subtractive panning or negative selection. This involves exposing the phage library to irrelevant proteins, carrier molecules, or closely related antigens before positive selection. This step helps remove clones that bind to common protein scaffolds or matrix components, thereby enriching for clones with true target specificity.
After several rounds of panning, it is important to analyze the pool of enriched clones. Sequencing a diverse set of phage clones helps identify whether certain sequences are being preferentially selected, which can indicate convergence toward high-affinity binders. This insight can guide clone prioritization for downstream validation and highlight structural motifs worth engineering.
Once promising binders are identified, they should be reformatted from their original scFv or Fab fragment into full-length IgG or other desired formats. This step is crucial because binding behavior can change upon reformatting. Validate these reformatted antibodies across key assays such as ELISA, Western blotting, and immunohistochemistry to confirm performance and specificity under real-world conditions.
Phage display remains a foundational technology for antibody generation, particularly when animal immunization is not viable or when in vitro control is preferred. Although newer methods like Plasma Cell Discovery (PCD) show promise with higher hit rates and in vivo maturation, phage display remains indispensable for early-stage research and rapid screening campaigns.
Need help planning your antibody discovery project? Explore how Boster Bio can support your antibody discovery project with phage display, hybridoma development, and plasma cell-based screening. Our team provides recombinant antibody expression and validation for IHC, ELISA, WB, and therapeutic workflows.
Contact us today to discuss your project.
Multiplex immunofluorescence, or multiplex IF, often looks “simple” on paper—until channels start bleeding into each other, weak targets disappear, or background forces you to crank exposure. In reality, most multiplex failures come from three upstream issues: channel planning, where brightness is mismatched to abundance, overlap, including spectral spillover and crosstalk, and missing controls, meaning no single-stain proof. The goal is not “more color,” but clean, interpretable signal with defensible imaging rules—so your colocalization reflects biology, not artifacts.
This post is a practical workflow for fluorophore selection in multiplex IF: plan channels by abundance, prevent bleed-through, and validate with controls. It’s written as a microscope-side SOP—decision rules plus checks—not a fundamentals-only overview. If you want to align upstream variables first, start here: sample preparation for IHC/ICC/IF and cell/tissue fixation.
To choose fluorophores for multiplex immunofluorescence and prevent bleed-through, (1) assign the brightest channel to the lowest-abundance target, (2) keep channels spectrally separated, (3) verify spillover with single-stain controls, and (4) optimize imaging settings per channel, then keep them constant within the same channel for any group/sample comparisons.
To make the intent clear: this post is a multiplex IF workflow—channel planning, single-stain spillover checks, and within-channel imaging consistency for comparisons. For fundamentals—what a fluorophore is, spectra basics, and selection basics— see how to choose the right fluorophore...

ELISA can be deceptively “clean.” The standard curve looks smooth, duplicates are close, and the plate reads without errors—yet the final concentrations don’t make biological sense, shift between runs, or fail to reproduce across days or operators.
In most cases, the issue isn’t pipetting skill. It’s experimental design: missing controls, standards that don’t match the sample range, dilution choices made without a quick pre-check, or plate reading/recording steps that ar
...
Transgenic animals are widely used in diagnostics, drug discovery, and cancer immunotherapy due to their ability to produce human-compatible antibodies. These genetically modified organisms are engineered to express fully human or humanized antibodies, providing a scalable and consistent platform for antibody
Western blot “quantification” is often treated like a software task. In reality, most wrong numbers come from two upstream issues: saturation (signal no longer scales with protein amount) and normalization (a reference that shifts or saturates). The goal is not “the darkest band,” but measurable signal in the linear range with a defensible reference—so your fold change reflects biology, not imaging artifacts.
This post is a practical, 5-minute workflow for western blot quantification: capture a quantifiable image, measure band intensity (densitometry), normalize (loading control or total protein), and calculate fold change. If you want broader context or need to fix blot quality first, these internal hubs are designed to be your next clicks:

Immunization is the first and most critical step in generating high-affinity antibodies. By introducing antigens into a host animal, researchers trigger an immune response that leads to the production of target-specific antibodies. The effectiveness of this process depends on several factors, including the choice of host species, antigen preparation, adjuvants, and immunization schedule.
This article outlines the most common immunization strategies used in antibody production and how these methods impact antibody diversity, affinity, and downstream applications.
Immunization is the starting point of all antibody development workflows. The primary goal is to introduce a foreign substance—called an antigen—into the host's body in a way that triggers a strong, specific immune response. This response involves the activation of B cells, which differentiate into plasma cells that produce antibodies targeting the antigen.
The strength and specificity of this first response set the stage for downstream antibody development, shaping critical fac
Antibody crosslinking is a process in which antibodies are used to link multiple antigens or molecules together. This interaction plays a significant role in various biological processes, experimental assays, and therapeutic applications, particularly those examining antibody-antigen interactions and protein interactions. While often intentional in research settings, crosslinking can also occur as a byproduct of fixation or monoclonal antibody design, influencing biological outcomes.
Antibody crosslinking occurs when an antibody binds to two or more targets at the same time, often pulling them into proximity—a mechanism that also contributes to B cell sorting and activation during antigen recognition. This interaction can occur naturally in biological systems, such as during immune complex formation, or be intentionally induced in the lab using secondary antibodies or chemical crosslinkers.
The resulting crosslinked structures help researchers study protein structures, amplify detection signals, and stabilize complexes for downstream analysis such as cross-linking mass spectrometry or structural modeling.
There are three main types of antibody crosslinking, each with distinct mechanisms and applications:
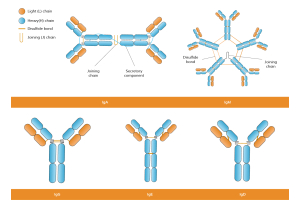
Antibodies are naturally bivalent (e.g. IgG) or multivalent (e.g. IgM), meaning they have two or more antigen-binding sites, structurally supported by intrachain and interchain disulfide bonds that stabilize their conformation. These sites can simultaneously bind identical or closely spaced antigens on the plasma membrane or within a protein complex, and their Fc regions can engage Fc receptors to mediate downstream immune responses. This physical bridging of molecules enables:
This results in:
Chemical crosslinkers are small molecules that form covalent bonds between proteins or between proteins and surfaces, often stabilizing protein structure for analytical or imaging purposes. Common reagents include:
Crosslinking is essential in many experimental workflows and biological processes because it helps localize targets, improve detection sensitivity, and enable selective bonding between specific chemical groups—an approach frequently paired with mass spectrometry for structural or functional analysis. Properly selecting the type of crosslinking ensures better assay design, preservation of protein structure, and reproducibility across applications.
Antibody crosslinking influences several key biological and experimental processes:
Crosslinking of cell surface receptors (e.g. CD3, CD28, Fc receptors) embedded in the plasma membrane by antibodies can activate intracellular signaling cascades. For example:
Naturally, crosslinking enables antibodies to form immune complexes with antigens. These complexes help:
In laboratory workflows, crosslinking amplifies signals:
Learn more about Western blot optimization guides on Boster Bio.
Learn moreDifferent antibody crosslinking methods are suited to specific assay formats like immunoprecipitation, Western blotting, and crosslinking antibodies for IHC. Below are two of the most commonly used approaches.
This method is widely used in IHC, ICC, WB, and ELISA. Secondary antibodies are designed to recognize and bind to primary antibodies. Because a single secondary antibody can bind to multiple epitopes on different primary antibodies, this process forms a branched or lattice structure that enhances signal output.
For example:
Applications include:
Chemical crosslinkers form covalent bonds between antibodies and their targets, or between proteins and solid supports, complementing the structural stability provided by natural disulfide bonds.
Common agents include:
Applications include:
Explore Boster Bio's antibody validation services to ensure your reagents perform as expected.
Learn moreWhile crosslinking is useful, it must be carefully controlled. Common concerns include:
| Implication | Description |
|---|---|
| Epitope Masking | Excessive crosslinking may hide antibody binding sites, especially in Formalin-Fixed Paraffin-Embedded (FFPE) samples. |
| Non-specific Aggregation | Over-crosslinked antibodies can stick to unintended targets or form clumps. |
| Reduced Binding Flexibility | Crosslinkers may alter antibody structure or overall protein structure, including disruption of disulfide bonds, which can lower binding affinity or limit epitope accessibility. |
| Signal Amplification | Proper crosslinking improves assay sensitivity and supports multiplexing. |
Antibody crosslinking has a wide range of applications:
To get the most reliable results for antibody crosslinking:
Need help selecting antibodies for IHC or WB? Browse Boster Bio’s complete antibody catalog.
Learn moreAntibody crosslinking is more than just a technical step—it’s a versatile tool that enables signal amplification, cellular signaling, B cell activation, immune targeting, and molecular detection. Whether you're analyzing receptor function or optimizing an immunoassay, understanding crosslinking’s mechanisms and implications helps ensure reliable, reproducible results. By selecting well-characterized antibodies and optimizing your data analysis, antibody affinity, and crosslinking conditions, you can unlock
How the Immune System Builds Its Defense
Antibody diversity allows the immune system to generate millions of unique antibodies, many of which can be developed through specialized antibody discovery and antibody generation services, each capable of targeting